Reports: ND654372-ND6: Molecular Simulation of Vapor-Phase Nucleation of Alkanes
 printer friendly
printer friendlyImpact
A doctoral student, Sai Pooja Mahajan, was involved with and fully supported by this project during the Spring Semester and Summer of 2015. This student graduated by the end of 2015 and has since joined Intel Inc. as a senior scientist.
Summary
The main goal of this project is to study via molecular simulation the homogeneous nucleation vapor-phase bubbles from metastable superheated alkane liquids, especially under negative pressures. Focus in alkanes in in part due to their relevance to petrochemical processes and products but also because, unlike studies for metastable stretched water, there are very few such studies for other liquids including alkanes. In fact, alkanes and mixtures of alkanes have been mostly investigated in the superheated regime through various experiments. The motivation behind studying the mixtures was to probe the effect of differences in volatility and chain-length (shape and size) on the nucleation process. Overall, we are also interested in investigating the spherical-bubble assumption of Classical Nucleation Theory for such systems.
During the eight months that a graduate student was involved with this project we were able to make progress in 3 fronts: (i) Setting up and test the methodology to use, (ii) application to a pure alkane system (n-hexane), (iii) application to a binary mixture (n-hexane and methane). While our results are still partial, simulations are in progress which will allow us to complete a first manuscript. In the following we detail the specific progress in those 3 fronts mentioned above.
1) Methodology implementation and testing.
As planned, the TraPPE united-atom forcefield was used to model hexane and methane in hexane and hexane-methane systems. To track the progress of the nucleation, the main order parameter “l” used to distinguish between the vapor and the liquid state during the transition was the volume of the largest bubble, W, which has been successfully used before in similar studies. It is noted that the algorithm for detecting “liquid-like” and “vapor-like” voxels is more nuanced in the case of non-spherical molecules (like hexane) and when two species exist (like in the hexane-methane mixture).
As planned, we used the Hybrid Monte Carlo (HMC) method in the isobaric-isothermal ensemble to simulate particular states (with given temperature and pressure) and the Umbrella Sampling (US) method to map out the free-energy barrier as a function of the order parameter l. In the runs used to generate preliminary results for the proposal, we used an in-house serial code to runs such HMC-US simulations for a Lennard-Jones systems. However, to allow for applications involving more complex molecules and larger systems, we have now implemented HMC-US with a more general, all-purpose, highly parallelizable freeware such as Lammps. Lammps is primarily a molecular dynamics engine and is not set up to readily handle the required HMC-US calculations. The HMC algorithm was hence implemented using a Lammps-python interface wherein a wrapper python script was used to call internal functions within Lammps to run short MD trajectories, calculate bubble volume and change the volume of the system. An additional program was written in Lammps to enact volume change moves to maintain constant pressure and the bubble volume calculation was implemented as a compute command within Lammps. The program was validated by reproducing HMC results reported previously for stretched and superheated Lennard Jones fluid.
In our US method the overall l range of interest was broken up into a number of boxes so that separate runs sample different boxes. Some overlap between neighbor boxes was allowed to improve the stitching of the box free energy profiles into a single curve.
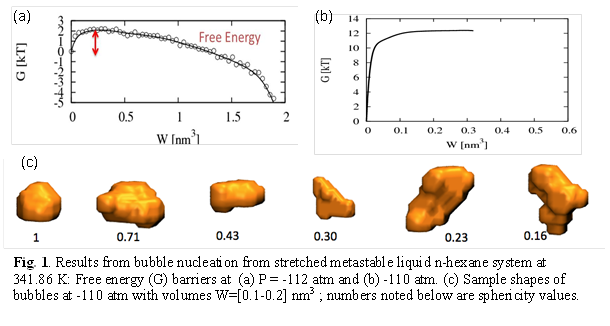
2) Results for pure hexane (Fig. 1)
The Hexane system was simulated for two types of deviation from the coexistence state at T=341.86K and P=1 atm: (i) The superheated metastable states with T= 430, 425 and 420K were chosen at P=1 atm, and (ii) the stretched metastable states with P=-112 atm and -110 atm at T=341.86K. In both cases, the limit for spontaneous nucleation was estimated when the barrier to nucleation was of the order of 1-2kBT; this happened at T=430 K and 1atm and at T=341.86K and -112atm. For the superheated system, the free energy barrier increased from 2 to 4 kBT as T changed from 430 to 420K. In contrast, for the stretched system a large change in barrier height from 1.5 to 11.5 kBT was observed for a small change in pressure from -112 to -110 atm.
The shape of the bubbles was seen to vary broadly for both cases and for all bubble sizes. For the stretched case, the shape distribution shifts towards a higher percentage of highly spherical bubbles as the bubble size grows while the opposite trend is observed for the superheated case. One reason for this difference could be that the system is at a higher temperature in the superheated regime where it is more prone to interfacial fluctuations. More results and analysis are needed to better quantify the above trends in free energy barriers and bubble shapes.
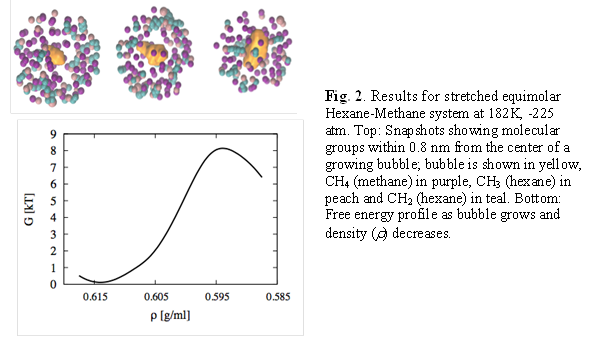
3) Hexane-Methane mixture (Fig. 2)
For equimolar mixtures of hexane and methane, there is only a narrow range of temperature where the two species can exist as a liquid; i.e., between 178 K (triple point of hexane) and 190.6 K (critical point of methane). We chose 184 K and 182 K to simulate the mixtures. Via multiple exploratory MD simulations we found that the system nucleates spontaneously at around -235 atm. Free energy barrier heights of 6 and 8 kBT were obtained for T, P=184 K, -230 atm and 182 K, -225 atm respectively. Our analyses so far reveal again the presence of non-spherical bubble shapes and a non-homogeneous population of hexane and methane molecules as a function of the radial distance from the center of the bubble: expectedly, the bubble cavity is predominantly surrounded by methane molecules. This phenomenon can be due to a thermodynamic driving force (partitioning of the move volatile species) and kinetic effects (disparity in molecular diffusivity) which we plan to uncouple via further analyses.