Reports: ND653061-ND6: PAH Excited State Dynamics
 printer friendly
printer friendlyPolycyclic aromatic hydrocarbons are a ubiquitous class of molecules, appearing in large abundance in crude oil, combustion products, and biomasses. Many of the chemical processes in their formation and reactions involve short lived excited states, which under the complex conditions of carbohydrate chemistry are often difficult to investigate in detail. In this project we study excited state dynamics of isolated PAHs in molecular beams. With this approach we study these molecules not only in the gas phase, with relevance to many combustion processes, but also under collision free conditions, to reveal their fundamental, intrinsic properties.
Three graduate students have been working on this project, Jacob Berenbeim and Faady Siouri, as well as a new graduate student, Samuel Boldissar, who was already involved last year as a senior undergraduate student. A number of undergraduate students have been involved with this work as well, exposing them to state-of-the art research in chemistry. Three of them have just graduated and, in part inspired by this experience, are now going to graduate school, Anthony Wong, Christopher Woods, and Adam Peters
(I) Picosecond spectroscopy
In the second year of this project we have developed a new line of research to study these excited states by picosecond spectroscopy. We have installed a new picosecond laser system in our laboratory.
The set-up consists of an Ekspla PL2251 picosecond Nd:YAG laser, pumping a PG401 optical parametric generator with a second harmonic generator (Fig 1). The system produces 10 picosecond pulses, continuously tunable from 193 nm to 2300 nm, with peak powers of the order of 50 MW. Part of the 266 nm Nd:YAG beam can be separated with a beamsplitter and delayed in mechanical variable delay stage for pump-probe experiments to measure excited state lifetimes.
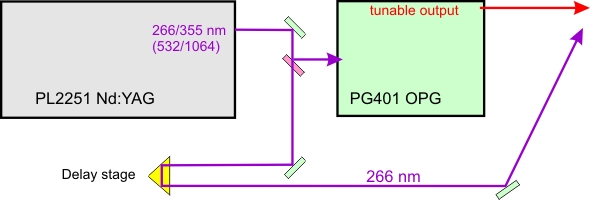
Figure 1
2-Aminopurine
To determine the capability of the new system, Figure 2 shows a pump-probe spectrum, we obtained for 2-aminopurine – a one ring compound. The excited state lifetime is 153 picoseconds.
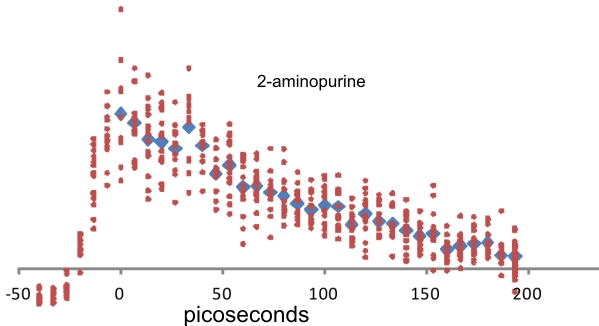
Figure 2
Cytosine
Figure 3 shows two one-color resonance enhanced multiphoton (REMPI) spectra for cytosine. We obtained the black trace with 10 nanosecond laser pulses (from a dye laser) and the blue trace with 10 picosecond laser pulses (from the OPG). In these spectra the molecule is ionized by two photons from the same laser. The laser is scanned and produces enhanced ion signal when the first photon is resonant with a vibronic transition in the molecule. If the excited state lifetime is significantly shorter than the laser pulse, the probability for absorbing the second photon diminishes. The excited state lifetime of the various peaks is indicated in Figure three in green. These lifetimes are deduced from the peakwidths obtained in high resolution. Excited states with lifetimes less than about 10 picoseconds cannot be ionized with the nanosecond laser. Those same levels can still be ionized with the picosecond laser without any problem. While the black spectrum essentially stops just above 32000 cm-1, the blue spectrum reveals the higher energy levels. This result is crucially important because we are studying the excited state dynamics of these compounds. Until now we were blind for the short lived excited states that are the most important for understanding the dynamics.
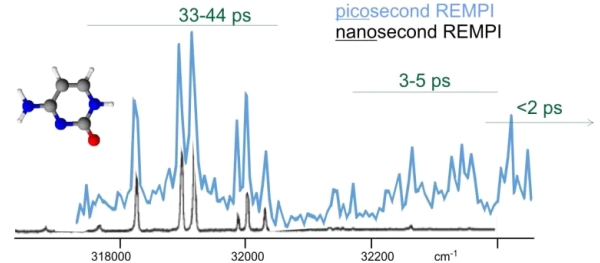
Figure 3
(II) Femtosecond spectroscopy
Adenine
The picosecond spectroscopy improves our time domain by three orders of magnitude while still providing small enough frequency bandwidth to perform vibrationally resolved spectroscopy (See Figure 3). This provides an ideal trade-off between pulse width (for observing dynamics) and bandwidth (for spectroscopy). However, for some aspects of our project it is also useful to have a capability in the femtosecond regime, which is another two orders of magnitude faster. Our department is acquiring a new femtosecond laser system, which will replace an older antiquated set-up. This is a shared facility and we have set up molecular beam apparatus in the same laboratory space. As preliminary work we have obtained a femtosecond pump-probe spectrum of adenine (shown in Figure 4) as well as perylene. Significantly, the new system will be tunable and have much better photon flux and stability, promising data with much better signal to noise. The preliminary work was important is setting up our experiment in the new facility and optimizing the approach.
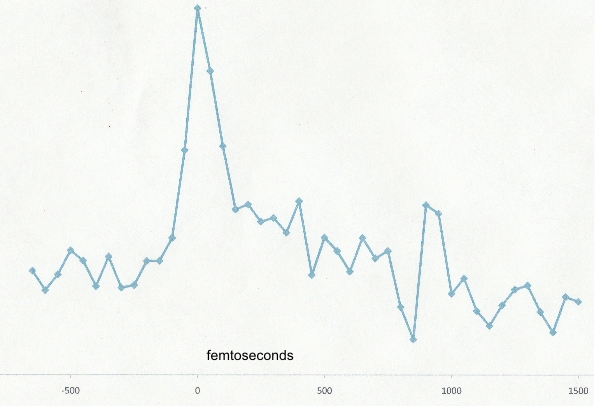
Figure 4
(III) Pyrimidine excited state dynamics.
This part of the work is continued from the first year. For pyrimidines UV absorptions leads to excitation to the S2 state. Next, subpicosecond de-excitation back to the ground state, S0, appears to be the dominant process. This process of internal conversion involves at least one conical intersection. However, a small portion of excited state molecules appears to populate a “dark state”, which we probe by ionizing with an excimer laser at 193 nm. We have obtained two-color two-photon ionization spectra for uracil (U) and thymine (T) as well as IR double resonant spectra, reported in the previous year period. As continuation of this study we have obtained pump-probe spectra to measure the lifetime of the dark state. This state is populated from a singlet excited state (S2) within picoseconds and then decays on the order of 50-70 ns for uracil and 200-300 ns for thymine. This lifetime is strongly dependent on the excitation wavelength by which S2 is first populated. In a surprising finding we observed that we can modulate the dark state lifetime by changing the ground state vibrational population prior to any electronic excitation. Figure 5 illustrates this result with pump-probe spectra for thymine. We obtain the blue spectrum with two UV pulses with a variable delay. To obtain the red spectrum we precede both UV pulse with an IR pulse, tuned to a resonant NH stretch frequency at about 2800 cm-1. The result is a reduction in the excited state lifetime of the dark state by almost one half. In our experiments the molecules are cooled in supersonic expansion to about 10K to ensure that all molecules are in the lowest vibrational state, to begin with. The IR excitation changes that vibrational distribution which in turn affects all subsequent dynamics.
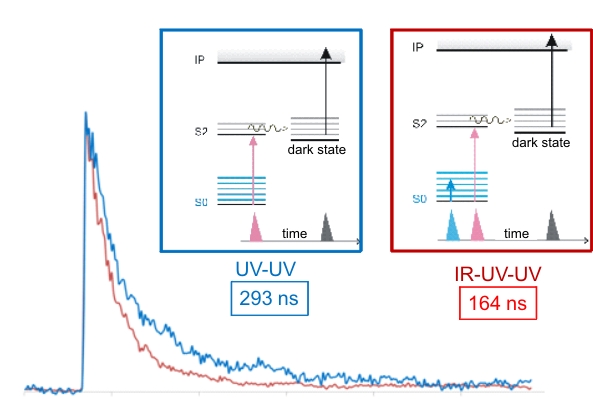
Figure 5










