Reports: ND354739-ND3: New Ligands for Catalysis Through Elaboration of the Phosphatriptycene Framework
Joseph P. Sadighi, PhD, Georgia Institute of Technology
The goal
of this project is the synthesis of sterically
encumbering, threefold-symmetric phosphines built
from the phosphatriptycene architecture using
selective directed metalation strategies. As a ligand
to transition metals, phosphatriptycene is known to
be a poor s-donor and a strong p-acceptor relative to triphenylphosphine,
because the geometric constraints on the system enforce unusually small angles
about phosphorus. Existing routes to phosphatriptycenes,
however, offer limited synthetic flexibility, and the supramolecular
architectures we seek have not been built from this scaffolding. In addition,
the routes to the parent ligand are synthetically demanding in ways that would
hinder our subsequent investigation. We therefore began with two lines of
inquiry, each pursued by one graduate student.
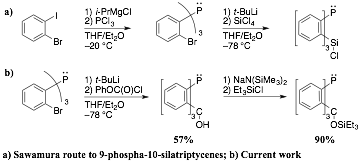
The
first line of inquiry consisted of a convenient synthesis of a phosphatriptycene derivative suitable for elaboration at
the aryl positions flanking the phosphorus. Here we have borrowed a key trick
from the work of Sawamura and coworkers on
9-sila-10-phosphatriptycenes: the use of tris(2-lithiophenyl)phosphine,
generated from tris(2-bromophenyl)phosphine, as the
key intermediate. Note that we could not simply start with Sawamura's
architecture, as we need the bridgehead atom in the 9-position to be smaller
than phosphorus, to make the intended pocket around the phosphorus large enough
to accept a metal atom. Therefore we closed the triptycene
framework using the phosgene equivalent phenyl chloroformate.
After protic workup, 9-hydroxy-10-phosphatriptycene
is obtained in 57% crystallized yield, on multigram
scale. For comparison, the elegant initial preparation of the parent phosphatriptycene by Bickelhaupt
and coworkers in 1974 requires five steps from commercially available
precursors, the last of which proceeds in 35% yield. The hydroxyl group,
present for synthetic convenience, presents a synthetic challenge, in that it
could direct metalation in positions adjacent to it,
rather than to the phosphorus. We therefore protected this group as a triethylsilyl ether in 90% yield.
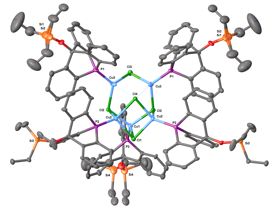
Before
proceeding toward the desired supramolecular
architecture, we prepared a copper(I) complex of the
9-siloxy-10-phosphatriptycene to measure key metrics by X-ray crystallography.
The copper(I) chloride complex of this ligand
crystallizes from a solution in dichloromethane / hexanes as a pentamer with bridging chlorides.
The
constrained geometry of this ligand relative to triphenylphosphine
is evident, for (Ph3P)CuCl
crystallizes as a chloride-bridged tetramer. Geometric calculations based on
this structure confirm that a pocket about the phosphorus, created by
replacement of flanking C–H bonds with C–O–aryl moieties,
should accommodate first-row transition metals readily.
The
elaboration of this ligand using directed metalation
has proved frustrating. We knew of no example in which a phosphine itself acts
as a directing group toward bases such as lithium amides or magnesium amides,
but hoped that reagents based on zinc or copper, both of which have a greater
affinity for phosphorus, would bind the phosphorus and deprotonate the nearest
C–H bond. Bases such as (2,2,6,6-tetramethylpiperidyl)zinc
chloride, lithium di(tert-butyl)(2,2,6,6-tetramethylpiperidyl)
zincate, and (2,2,6,6-tetramethylpiperidyl)copper(I), however, showed no
reaction with the 9-siloxy-10-phosphatriptycene. The metalation
of phosphine oxides by very strong bases such as tert-butyllithium is known, and
we were able to oxidize the phosphatriptycene to the
corresponding phosphatriptycene oxide cleanly, but
our substrate gave rise to a complex mixture of ring-opened products under
these conditions. Less reactive magnesium amide bases, disappointingly, gave no
reaction at all. In view of the affinity of zinc and copper for sulfur, we
prepared the phosphatriptycene-P-sulfide and treated it with the zinc and copper amide reagents,
but again observed no reaction.
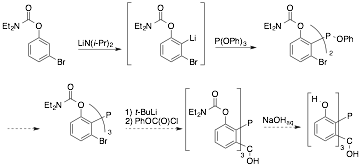
We are
currently investigating a modified strategy, in which the positions adjacent to
phosphorus are already functionalized before the triptycene
framework is assembled. The directed lithiation of
3-bromo-1-(N,N-diethylcarbamoyl)phenol by lithium diisopropylamide
has been reported. We are attempting to use this aryllithium
to prepare the corresponding triarylphosphine. At
present, we have succeeded in placing two of these aryl groups on phosphorus,
as judged by 31P NMR spectroscopy and mass spectrometry; the third
P–C bond formation remains difficult, possibly due to steric encumbrance.
We cannot heat the reaction to drive it to completion, as the aryllithium undergoes lithium bromide elimination to form a
reactive benzyne. We remain confident that conditions
can be found to assemble this new triarylphosphine.
With it in hand, we could readily assemble a phosphatriptycene
with flanking hydroxy groups, the arylation
of which should be feasible.
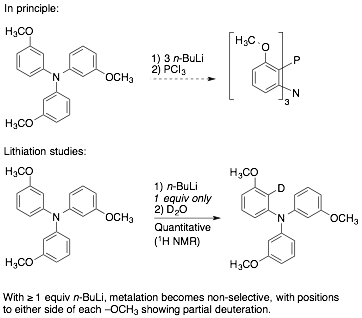
Finally,
we sought to synthesize 9-aza-10-phosphatriptycenes, believing that directed metalation of triarylamines could
likewise offer a convenient route to an already-elaborated architecture, one
that could be incorporated before the triptycene was
assembled. We chose the methoxy substituent for a
first pass. Tris(3-methoxyphenyl)amine is a known compound, readily prepared
in high yield in a single step from commercially available materials by
Buchwald-Hartwig coupling. A methoxy
group is one of the best known directing groups for arenelithiation, and we knew
that directed lithiation normally proceeds between
two directing groups if they are present in a meta-arrangement. We did not know, however, whether the poorly
basic triarylamine nitrogen could serve as the second
directing group, to achieve the desired geometry. We learned that it does so
quite effectively for a single metalation, but that
attempted triple metalation gives rise to loss of
selectivity.
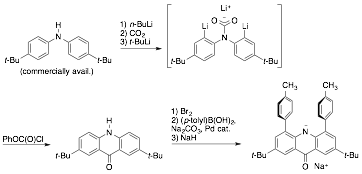
Building
on our findings in directed lithiation, however, we
have achieved the synthesis of an acridone, not
previously known, from a diarylamine. The use of a carbamate anion as a removable directing group was known
for the single lithiation of N-alkylanilines, but we did not know
whether it would work cleanly for a double lithiation.
Using this route, we have developed a multigram-scale
synthesis of a previously unknown acridone, and have
gone on to elaborate this framework into what we hope will be a useful ligand.
Although very different than the phosphatriptycenes
we set out to make — this is a nitrogen-based s-donor with negligible p-accepting characteristics — this
diarylated acridone offers an array of new directions as a ligand in its own
right.
 printer friendly
printer friendly