Reports: DNI653482-DNI6: Signature of Molecular Environment in Spectroscopy Measurement of Water and Aqueous Solutions Studied by Advanced ab Initio Methods
 printer friendly
printer friendlyResearch report for the second year 2014-2015 for PRF grant support
By combining density functional theory (DFT) for the electrons and classical dynamics for nuclei, ab initio molecular dynamics (AIMD) allows the direct model of complex H-bonded liquids at finite temperature without relying on empirical force fields. In AIMD, the nuclear dynamics and the adiabatic evolution of the DFT ground state are treated at the same footing. Therefore, it enables an explicit description of the changes of electronic orbitals during the H-bond fluctuations. It is well-known that the most widely used XC functionals with the generalized gradient approximation (GGA) has severe limitations when applied to water. Most notably, GGA-DFT suffers from the well-known self-interaction error which leads to excessive proton delocalization, yielding red shifts in the O-H stretching frequencies and a severely over-structured H-bond in predictions. A commonly adopted method to alleviate the self-interaction error is the application of hybrid XC functional, in which a fraction of exact exchange energy is mixed in GGA functional approximation. However, hybrid XC functional is extremely computational expensive and has been so far only rarely seen in liquid simulations. In addition, GGA also ignores the nonlocal electron correlation effects originating the van der Waals (vdW) interactions that are crucial in making water denser than ice. Finally most AIMD simulations of H-bonded liquids so far have adopted classical mechanics for the nuclei. However, the light atom (Hydrogen) deviates from classical behavior significantly, even at room temperature. Treating the quantum nuclei effect requires the Feynman discretized path integral (PI) scheme [19-21], but for reasons of computational cost this technique has been rarely applied.
Fig. 1: (a) The oxygen-oxygen structure factors, SOO(Q), of liquid water obtained from PBE0 + TS-vdW AIMD simulation and various X-ray scattering experiments. (b) The oxygen-oxygen radial distribution functions, gOO(r), of liquid water from PBE0+vdW-AIMD simulation and various scattering experiments. (c) The chlorine–oxygen and (d) chlorine–hydrogen radial distribution functions, gClH(r) and gClO(r), of the aqueous chloride ion solution.
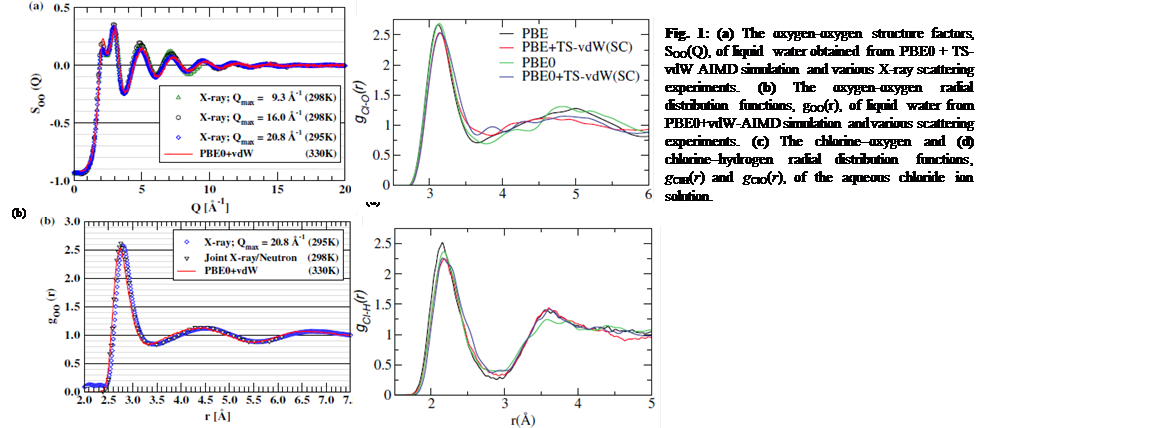
In order to address the above issues, during the first year of the PRF support (2013-2014), we have first performed simulations of ambient liquid water using a hierarchy of exchangecorrelation (XC) functionals to investigate the individual and collective effects of exact exchange (Exx), via the PBE0 hybrid functional, non-local van der Waals/dispersion interactions, via a fully self-consistent density-dependent dispersion correction, and an approximate treatment of nuclear quantum effects, via a 30 K increase in the simulation temperature, on the microscopic structure of liquid water. Based on these AIMD simulations, we found that the collective inclusion of Exx and vdW as resulting from a large-scale AIMD simulation of (H2O)128 significantly softens the structure of ambient liquid water and yields an oxygen-oxygen structure factor, SOO(Q), and corresponding oxygen-oxygen radial distribution function, gOO(r), that are now in quantitative agreement with the best available experimental data. This level of agreement between simulation and experiment demonstrated herein originates from an increase in the relative population of water molecules in the interstitial region between the first and second coordination shells, a collective reorganization in the liquid phase which is facilitated by a weakening of the hydrogen bond strength by the use of a hybrid XC functional, coupled with a relative stabilization of the resultant disordered liquid water configurations by the inclusion of non-local vdW/dispersion interactions. This increasingly more accurate description of the underlying hydrogen bond network in liquid water also yields higher-order correlation functions, such as the oxygen-oxygen-oxygen triplet angular distribution, POOO(θ), and therefore the degree of local tetrahedrality, as well as electrostatic properties, such as the effective molecular dipole moment, that are in much better agreement with experiment.
Fig. 2: Total density of states (DOS in red) and real-space projected density of states (PDOS in blue) for the aqueous Cl− ion solution computed using the hybrid PBE0 XC functional based on structures (configurations) generated from AIMD trajectories at the (a) PBE, (b) PBE+TS-vdW(SC), (c) PBE0, and (d) PBE0+TS-vdW(SC) levels of theory. The DOS and PDOS above were obtained from electronic structure calculations on 200 configurations taken from each AIMD trajectory.
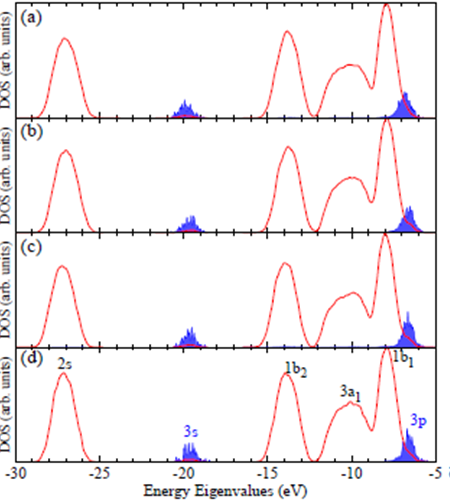
By obtaining the very important corrections on the structure of H-bond network, during the second year (2014-2015) of the PRF support, we started to apply the accurate H-bond simulations onto the ion solutions of biological importance which include Cl- and so-called Hofmeister series (such as Ca2+, Mg2+, Li+, Na+, K+, F-, Cl-, Br-), solutions in liquid water. In particular, the solvation and electronic structure of the aqueous chloride ion solution was investigated using DFT based AIMD. From an analysis of radial distribution functions, coordination numbers, and solvation structures, we found that Exx and non-local vdW interactions effectively weaken the interactions between the Cl− ion and the first solvation shell as shown in Fig. 1 (c) and (d). With a Cl-O coordination number in excellent agreement with experiment, we found that most configurations generated with vdW-inclusive hybrid DFT exhibit 6-fold coordinated distorted trigonal prism structures, which is indicative of a significantly disordered first solvation shell. By performing a series of band structure calculations on configurations generated from AIMD simulations with varying DFT potentials, we found that the solvated ion orbital energy levels (unlike the band structure of liquid water) strongly depend on the underlying molecular structures. In addition, these orbital energy levels were also significantly affected by the DFT functional employed for the electronic structure; as the fraction of Exx was increased, the gap between the highest occupied molecular orbital of Cl− and the valence band maximum of liquid water steadily increased towards the experimental value as shown in Fig. 2 (a-d).
Currently, we are applying the above advanced H-bond modeling onto more broadly the Hofmeister series (such as Ca2+, Mg2+, Li+, Na+, K+, F-, Cl-, Br-) solutions. We are planning to finish the computational simulations of Ca2+, Na+, K+ solutions and write, submit the manuscripts during the requested extension of the funding support to the third year.










