Reports: ND154228-ND1: A Metathesis-based Synthesis of Dodecahedrane, isomers and analogues thereof
 printer friendly
printer friendlyOur Efforts to Synthesize Triquinacene and Dodecahedrane.
In this grant we sought to exploit the remarkable Z-selectivity of recently developed olefin metathesis catalysts to achieve practical syntheses of triquinacene (2) and dodecahedrane (5) using the synthetic strategy outlined in Scheme 1. In the first step of this synthesis, hexaene (1) would undergo ROM/RCM to generate triquinacene in situ; subsequent dimerization and two sequential isomerizations (3→4) would produce dodehedrane.
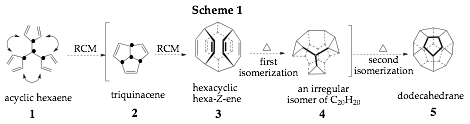
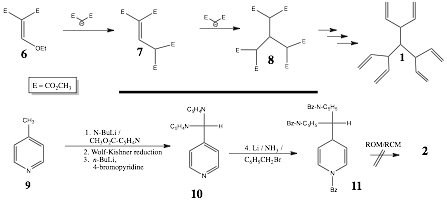
In the first year of this two-year grant, we learned that while hexaester 8 could be efficiently prepared from 3-ethoxymethylenedimethyl malonate (6), it's conversion to acyclic hexaene (1) has thus far eluded us (Scheme 2). An alternative strategy was studied whereby 4-methylpyridine (9) was converted to the tris-pyridinylmethane 10 in good overall yield. Surprisingly, although the Birch reduction of 10 proceeded in high yield and cleanly, the trisenamine intermediate 11 did not undergo olefin metathesis, regardless of the catalyst, or the reaction conditions employed. Moreover, intermediate 11 was very sensitive to hydrolysis. Model substrates, such as hexa-1,5-diene, underwent straightforward ROM/RCM, thereby suggesting this may be a previously unknown limitation of ROM/RCM reactions. Attempts were also made to prepare triquinacene (2) via two published routes. However, as yet significant quantities of triquinacene have not been produced to permit the investigation of the dimerization and proposed isomerizations.
Scheme 2
In May of 2015, Tom Irvin left UGA to join Dr. Kenner Rice at the National Institutes of Health as a postdoctoral research assistant. This Fall I have taken on a new student to continue this investigation. I have avoided conducting this research using undergraduate researchers as, despite my personal supervision, working with alkai metals, large-scale Birch reductions, and other transformations severely tests their skills and our laboratory safety measures. While we have made some progress on the work which was funded, significant progress was made by Charles Stanton, III, a third year doctoral candidate, on a collaboration with computational chemists (UGA), and inorganic chemists (Princeton University and University of California at San Diego) which also represented another significant and new direction in my research program. Charles has been working on this project for two years now, which predates both the submission of my proposal and the funding of this grant. A summary of the past year of Charles' accomplishments using PRF funding follows.
Catalytic CO2 reduction.
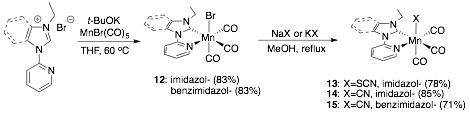
The synthesis, electrochemical activity, and relative photodecomposition rate of four new Mn(I)-N–heterocyclic carbene complexes were reported: [MnX(N-ethyl-N′-2-pyridylimidazol-2-ylidine)(CO)3] (X = Br, NCS, CN) 12, 13, and 14 and [MnCN(N-ethyl-N′-2-pyridylbenzimidazol-2-ylidine)(CO)3] 15 (Scheme 3). All four compounds displayed an electrocatalytic current enhancement under CO2 at the potential of the first reduction, which ranged from -1.53 V to -1.96 V versus the saturated calomel electrode (SCE). Catalytic CO production is observed for all species during a four-hour preparative-scale electrolysis, but substantial H2 was detected in compounds where the axial ligand, X, was not Br. All species eventually decomposed under both 350 nm and 420 nm light, but complex 15 (X = CN) lasted up to 5 times longer under 420 nm light as a result of a blue-shifted MLCT band.
Scheme 3
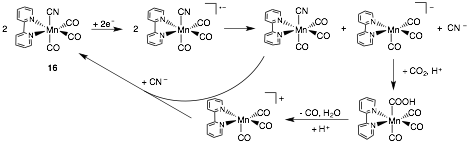
In a model study, we substituted the axial bromide for a nitrile which yielded Mn(CN)(2,2'-bipyridine)(CO)3 (16, Scheme 4); the electrocatalytic ability of complex 16 was then established. In particular, this structural modification shifts the first and second reductions to more negative potentials (−1.94 and −2.51 V versus Fc/Fc+, respectively), but imparts quasi-reversibility at the first feature. The two-electron, two-proton reduction of CO2 to CO and H2O is observed at the potential of the first reduction. Data from IR spectroscopy, electrochemistry, cyclic voltammetry, and controlled potential electrolysis indicates that this behavior arises from the disproportionation of two one-electron reduced species to generate the catalytically active species. Computations using density functional theory support this new mechanism. Future work will focus on structural modifications that produce more efficient catalysts.
Scheme 4
Word Count 729










