Reports: ND753456-ND7: Supramolecular Gels and Nanocomposites with Multiple Stimuli Responsive Behavior
 printer friendly
printer friendlyStimuli-responsive polymers, also known as “smart polymers”, are materials that undergo large changes in their properties (e.g., color, shape, etc.) in response to stimuli, such as temperature, light, etc.1–3 Stimuli-responsive polymers could find application in actuators, drug delivery systems, and self-healing materials.4–7 Stimuli-responsiveness is usually driven by modulating chemical and physical interactions such as π–π bond interactions, which refers to noncovalent bonding associated with nearby π electron containing functional groups such as aromatics.
A major goal of our research was to study pyrene end-functionalized telechelic polymers that respond rheologically to a thermal stimulus by modulating π–π interactions of the end-groups. For typical polymer molecular weights (i.e., 1–100 kg mol-1), the pyrene chain ends are expected to represent less than 1 wt% of the host polymer repeat units as summarized in Table 1.
Table 1. Comparison of poly(dimethylsiloxane) (PDMS) precursor properties provided by the manufacturer to the measured/calculated properties. N100 represents the precursor amine terminated PDMS used to functionalize pyrene end labeled PDMS.
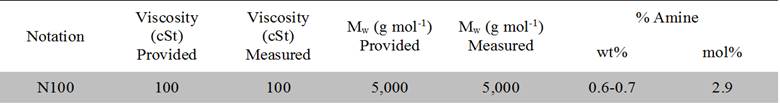
In this research, amino-propyl terminated PDMS (denoted N100) was used as a starting material, and the end groups were fully pyrene functionalized (denoted P100). This introduces the formation of pyrene nanocrystals and associated physical crosslinks as shown in Figure 1. As a result of this structuring, P100 formed solid gels at room temperature. The driving force for producing nanocrystals is the formation of complementary π–π interactions with a binding energy of 20–26 kJ mol-1, which is far above thermal energy at room temperature (kT ~2.4 kJ mol-1).
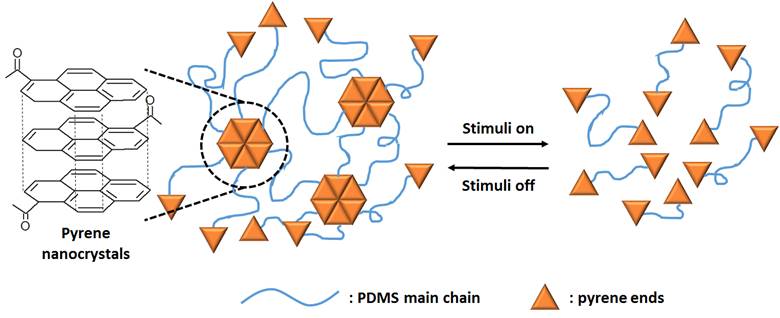
Figure 1. Schematic showing structuring and stimuli responsive behavior of pyrene end-functionalized PDMS.
To explore stimuli-responsive capabilities, temperature was modulated through various heating and cooling cycles while measuring the rheology of the materials. By including an isothermal annealing step in every thermal cycle (horizontal line in the blue curve), complete recovery of the rheological properties was observed over repeated cycles (Figure 2). This material oscillated from solid-like gels (G’ > G”) to viscous liquids (G” > G’) depending on the temperature. Considering the very large rheological changes in response to temperature and complete reversibility, such materials could be used effectively as heat sensors, repositionable adhesives or in tunable damping systems.
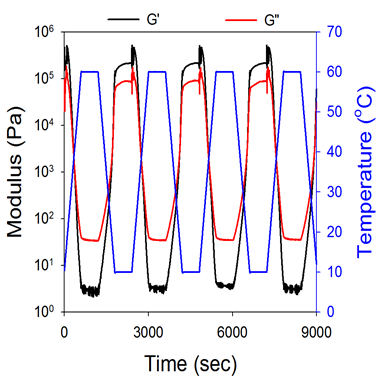
Figure 2. Dynamic moduli during temperature cycling showing complete reversibility when thermal annealing steps are incorporated into every thermal cycle. Black and red curves represent G’ and G”, respectively, and the blue curve represents the temperature profile.
Besides the large change in rheological properties stimulated by moderate heat, the thermal properties of pyrene end-functionalized PDMS are tunable by the incorporation of pure PDMS or pyrene dye additives as shown in Figure 3. Addition of 20 wt% pure PDMS diluted the density of the pyrene ends in the system, which correspondingly reduced the pyrene melting temperature (Figure 3a). The addition of 5 wt% free pyrene increased the likelihood of preferential interactions of the pyrene chain ends of P100 with free pyrene, which decreased the possibility of pyrene end-group nanocrystal formation. In this case, the free pyrene is more mobile than the pyrene attached to chain ends and can effectively “shield” π–π interactions between adjacent pyrene chain ends.
While the thermal properties of P100 were modified with polymeric and small molecule additives by eliminating pyrene nanocrystal formation, graphene oxide (GO) nanoparticle additives appeared to result in a beneficial enhancement in the thermal properties as shown in Figure 3. GOs, having an abundance of double bonds and π electrons, can promote pyrene end groups to bond with their surfaces; therefore, incorporating 1 wt% GOs (0.44 vol%) resulted in an increase of melting temperature from 42 to 88.8 °C.
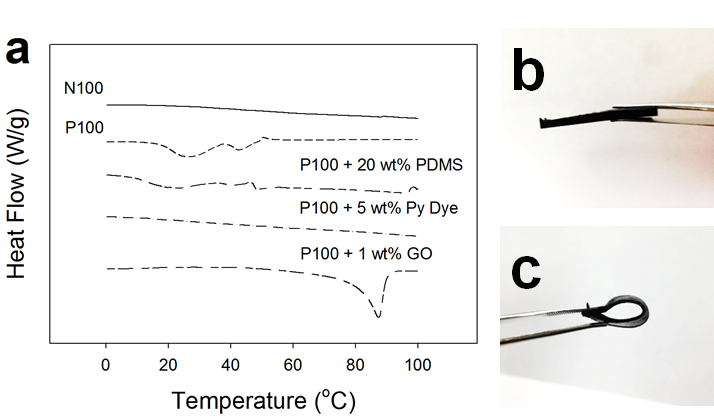
Figure 3. (a) DSC thermograms collected upon second heat for N100 precursor and P100 with pure PDMS, free pyrene (Py Dye) or GO. (b) Self-supporting bar sample composed of P100/1 wt% GO and its (c) flexibility.
It was possible to cast this P100/GO composite into a bar using a mold as shown in Figure 3b and c. The composite was free standing (Figure 3b) and flexible (Figure 3c), which demonstrated the presence of robust π–π interactions. Such materials could find application as tunable damping materials, heat or light sensors, conductive gels, or light repositionable adhesives.
The American Chemical Society–Petroleum Research Fund has been critical for research not only to this project, but it has also spawned several other entirely new small projects. This grant has primarily supported one full time graduate student who mainly worked on this study for their doctoral degree. This has provided them an ability to deepen their understanding of polymer synthesis and characterization, and hone their skills with rheology and other scientific instrumentation. The student published 1 paper on the topic of stimuli-responsive polymers and presented 1 poster presentation and 1 oral presentation.8
References
1. R. J. Wojtecki, M. A. Meador, S. J. Rowan, Nat. Mater. 2011, 10, 14–27.
2. E. M. White, J. Yatvin, J. B. Grubbs, I. I. I. J. A. Bilbrey, J. Locklin, J. Polym. Sci., Part B: Polym. Phys. 2013, 51, 1084–1099.
3. H. G. Borner, H. K€uhnle, J. Hentschel, J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 1–14.
4. E. Cabane, X. Zhang, K. Langowska, C. G. Palivan, W. Meier, Biointerphases 2012, 7, 1–27.
5. P. Cordier, F. Tournilhac, C. Soulie-Ziakovic, L. Leibler, Nature 2008, 451, 977–980.
6. T. F. A. De Greef, M. J. Kade, K. E. Feldman, E. J. Kramer, C. J. Hawker, E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem. 2011, 49, 4253–4260.
7. D. Chen, J. Yoon, D. Chandra, A. J. Crosby, R. C. Hayward, J. Polym. Sci., Part B: Polym. Phys. 2014, 52, 1441–1461.
8. H. Ha, K. Shanmuganathan, Y. Fei, and C. J. Ellison. J. Polym. Sci., Part B: Polym. Phys. 2015, DOI: 10.1002/polb.23805.










