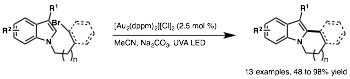The conversion of solar energy into chemical energy in
photosynthesis has enthralled scientists for decades. Sunlight could be used as an inexpensive, green, and sustainable source of
energy to induce the application of photochemistry in synthesis is therefore
restricted.In response to this
limitation, several light- absorbing photocatalysts
have been developed in an effort to mimic the light-harvesting abilities of bio-complexes
found in natural processes, such as photosynthesis. Increased attention is being placed on
visible-light-mediated photoredox processes for the development of efficient
and waste-minimizing processes of general applicability that should decrease
our dependence on toxic chemical products. To mimic Nature's photosynthesis, chemists
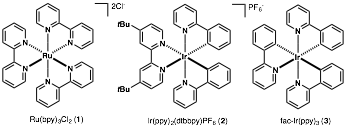
have developed a variety of photoredox complexes. Among theses, polypyridineRu(II)
and Ir(III) complexes (1-3) possesses high-energy,
long-lived and highly emissive excited states to be useful in organic synthesis
(Figure 1). Although these photoredox catalysts (1-3) are able to cleave
C-X bonds either by direct reduction from the catalyst excited state (oxidative
quenching) or via two single electron transfer (SET) processes (reductive
quenching), their reduction potentials (-1.31 V to -1.73 V versus SCE) limit
the scope of the radical intermediates to precursors having activated or weak
C-X bonds such as bromomalonate, electron deficient
benzyl bromides, polyhalomethanes and C-I. Typically, methods to generate carbon-centered
radicals from unactivated C-X bonds require the use
of hazardous radical initiators such as AIBN and Et3B, and toxic organotin reagent as a source of hydrogen.
Figure 1
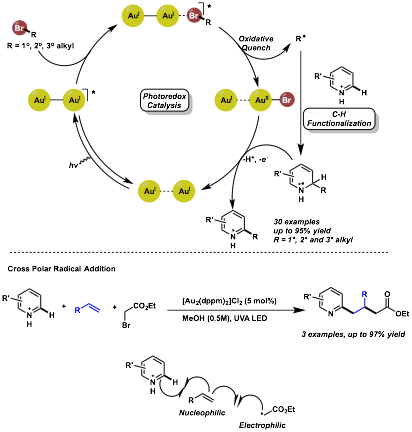
In late 2013, our group reported a UVA-enabled reductive radical
reaction of unactivated alkyl and aryl bromides in
the presence of a dimeric phosphine gold complex [Au2(dppm)2]Cl2 as the photocatalyst
(Figure 2).UVA irradiation of [Au2(dppm)2]Cl2
generates long-lived excited states which can undergooxidative
and reductive quenching cycle. It
was demonstrated that the applicability of this photoredox process is not
limited to intramolecular processes, but that intermolecular transformations
are also possible. Although the
mechanism was not completely understood, our results demonstrated the uniqueness
of this transformation and the high synthetic potential.
Figure 2
Progress report
The goal of this grant is to accelerate the development of photoredox catalysis as an
effective strategy for the construction of carbon-carbon bonds.The use of photoexcited
gold dinuclear complexes to generate radicals from unactivated carbon-halogen bonds provides a new plateform to perform radical reactions and design new
transformations. Early 2015, we
reported a new protocol for the reductive deoxygenation
of primary alkyl alcohols (Figure 3-A).
This method takes advantage of the unexpected light-mediated bromination with CBr4 in DMF using UVA-LED and
its compatibility with the photoredox process. The high synthetic value of this
transformation was demonstrated through the formation of alkyl radicals leading
to reduction (deoxygenation) and cyclization
products. Taking
advantage of the formation of the Vilsmeir-Haack
reagent, we wanted to apply the photogeneration of Vilsmeier-Haack reagent in a one-pot procedure for the
synthesis of amides from readily available carboxylic acids and amines.As shown in Figure 3-B, this new process
was conveniently applied to a simple one-pot synthesis of amides. Several amides and anhydrides were
obtained in high yields. This study
demonstrates the importance of photochemistry in developing fast, efficient and
reagent minimizing methods in organic synthesis.
Figure 3
Later in 2015, we reported the use of photoredox catalyst [Au2(dppm)2]Cl2
for the generation of organic free radicals in the functionalization of indoles (Figure 4).Operating through an oxidative quenching cycle, this method proves to be
highly efficient for the synthesis of substituted indoles. This process allows for simple access of
carbon-centered radicals not attained with popular photoredox catalysts and
without the use of harsh conditions and toxic reagents.
Figure 4
Part of the current PRF proposal, we have described the development
of photoredox Minisci reaction.Radical heteroaromatic
additions like the Minisci reaction are important
transformations for the chemical industry. We are pleased to report that the generation
and addition of nucleophilic alkyl radicals to heteroarenes from unactivatedbromoalkanes has been achieved for the first time on a photocatalytic
platform. We are currently
developing a universal and efficient Csp3-Csp2 coupling strategy for the
direct C-H alkylation of heteroarenes using a dimeric
gold(I) photoredox catalyst, [Au2(dppm)2]Cl2 (Figure 5). At the present time, we have produced
more 30 examples in high yields ranging from 50 to 95% which
include cross-polar radical addition.
Figure 5
Future Plans and Outlook
In addition to developing new synthetic methods, we are currently
establishing a thorough mechanistic investigation to determine the mode of
action of the photoexcited dimeric gold complex.In other words, does the oxidative or
reductive quenching cycle operate in an inner or out-of-sphere mechanism? Answer to these questions will not only
be fundamental but help with the design of new dimeric
gold complexes for specific reactivity to transform petroleum feedstock to
important molecules.
 printer friendly
printer friendly