Reports: ND453383-ND4: A New Spectroscopic Method to Assess the Oxidative Capacity of Iron Complexes in Catalytic Oxidation Reactions
 printer friendly
printer friendlyNARRATIVE
1. Approach. Using our dye assay of reactivity, we compared a set of iron catalyst (Fig. 1) that differ in their architecture with respect to ligand denticity, and number of open coordination sites.
We utilized malachite green (MG, Fig. 11) because i) its major absorption maximum at 618 nm minimizes overlap with absorption of iron complexes; ii) the oxidation products of MG are known and colorless;1 iii) in most cases, MG was completely degraded, facilitating comparisons.
Even though at least a few of the complexes can facilitate synthetic oxidation of MG with low catalyst load, optical concentrations that obey Beer’s Law are necessarily lower and the reactions consequently slower. Such conditions necessitated complex concentrations in excess of MG concentration. While these conditions do not provide turnover numbers with MG, they are still relevant and informative because they reflect reactivity.
The reaction(s) of the iron complexes with MG and H2O2 were followed by time-dependent changes in spectra and, in the case of MG, in a single wavelength kinetic mode at 618 nm. For all but [Fe(OTf)2(NNON)], in the presence of H2O2 MG absorption is lost to over 90% (Fig. 2). Like other similar reactions,1 the data exhibit pseudo first order behavior.
To understand the concentration dependence of the reaction on the metal complex and permit a way of comparison that accounts for the iron concentration, reactions with MG were conducted as a function of complex concentration.2 The resulting slope in mM-1min-1 is similar in form to a second order rate constant that demonstrates that the rate constants reflect iron complex reactivity (since kobs depends on iron complex concentration)2 (Fig. 2, Table 1). kobs at 25 mM for all the complexes was extrapolated from the concentration dependences.
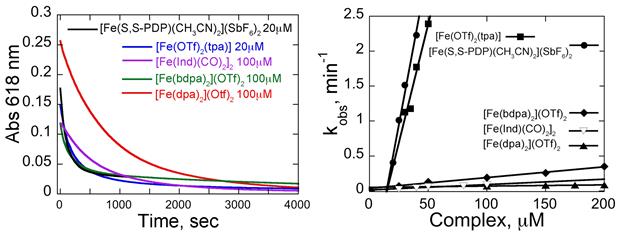
Fig. 2 (Left). Traces of the loss of 1 mM MG absorbance at 618 nm. (Right) Complex concentration dependence of kobs. Conditions: 1 mM MG, 0.06% H2O2, CH3CN, 25 °C.
Table 1. Complex Reactivitiesa
|
|
kobs (min-1) at 25 mM |
Concentration Dependence kobs mM-1min-1 | |
|
Complex |
|
H2O2 |
tBuOOH
|
|
[Fe(S,S-PDP)(CH3CN)2](SbF6)2 |
0.98
|
9.0e-2±1.3e-2 |
6.2e-3±2.5e-4
|
|
[Fe(OTf)2(tpa)] |
0.70
|
6.0e-2±7.0e-3 |
6.9e-2±7.0e-3
|
|
[Fe(bdpa)2](OTf)2 |
0.077
|
1.7e-3±1.2e-3 |
c |
|
[Fe(dpa)2](OTf)2 |
0.059 |
1.1e-3±9.5e-4 |
3.0e-6±2.7e-5 |
|
[Fe(Ind)(CO)2]2 |
0.033 |
8.7e-4±1.6e-4 |
c |
|
[Fe(OTf)2(NNON)] |
0.40
|
≈ 0c |
c |
a1 mM MG, 0.06% peroxide in CH3CN, 25 °C. bExtrapolated from concentration dependence. cMG not appreciably degraded.
The following insights were obtained:
1. The most active complexes are [Fe(S,S-PDP)(CH3CN)2](SbF6)2 and [Fe(OTf)2(tpa)]. Both have two open cis coordination sites, confirming a predicted important SAR.3 The remaining complexes, which do not share this feature, are at least 35-fold weaker. Thus one SAR is established.
2. While most complexes supported slower reaction rates with tBuOOH than H2O2 (consistent with the former being a poorer oxidant4), for [Fe(OTf)2(tpa)], the concentration dependences are the same for both tBuOOH and H2O2, suggesting a difference in mechanism relative to the other complexes.
3. For the two most reactive complexes, fast-forming isosbestic points in iron complex spectra indicate the formation, in situ, of a single species at least 10-fold faster than the degradation of MG, consistent with a faster reaction of the iron complexes with H2O2 to form another single species prior to MG oxidation.
4. [Fe(OTf)2(NNON)] displays no appreciable concentration dependence; serial addition of the complex5 was necessary to achieve complete MG degradation.
2. Turnover and Other Dyes. We surveyed other dyes to: i) find a dye that is oxidized in the presence of catalytic amounts of complex under spectroscopic conditions so turnover can be studied. ii) test if reactivities are correlated with the redox potential of the dye substrate. Preliminary studies with 13 dye substrates yielded a group that could be completely oxidized at 1:1 stoichiometry (trypan blue, brilliant blue FCF, Evans blue, indigo trisulfonate (ITS)). ITS (Fig. 1) is most vulnerable and can be oxidized at 20% catalyst load. There is no obvious correlation between redox potential and reactivity.
These preliminary results demonstrate that our approach provides quantitative information regarding relative metal complex reactivities and will be leveraged for further studies.
3. Impact. One graduate and one undergraduate have been trained in spectroscopic techniques needed for the above measurements and each have presented posters at regional events. One graduate student prepared the complexes needed for the studies. We have prepared a manuscript that is currently in revision, and are currently preparing a revision of an NSF grant application.
4. References.
(1) Oturan, M. et al. J. Appl. Cat. B 2008, 82, 244.
(2) van Eldik, R. et al. Inorg. Chem. 2008, 47, 2994.
(3) Que, L. et al. J. Am. Chem. Soc. 2002, 124, 3026.
(4) Loach, P. A. In CRC Handbook of Biochemistry; 2nd ed.; Sober, H. A., Ed.; CRC Press: Cleveland, 1973.
(5) Collins, T. J. et al. Chem. Eur. J. 2015, 21, 6226.










