Reports: DNI154405-DNI1: Direct Carbon-Carbon Bond Forming Reactions with Hypervalent Iodine Reagents: Redefining Formal SNAr Reactivity with Carbon Nucleophiles
 printer friendly
printer friendlyDiaryliodonium salts are latent precursors of aryl electrophiles, aryl radicals, and arynes. This diverse reactivity in metal-free reactions has led to a renaissance in synthetic methodology; symmetric diaryliodonium salts are routinely used in many modern methods. In our ACS PRF proposal we outlined a strategy to study these reagents as aryl electrophiles in direct carbon-carbon bond forming reactions, without the need for exogenous metal catalysts. Specifically, we proposed to expand formal nucleophilic aromatic substitution (SNAr) reactivity by engaging diaryliodonium electrophiles in reactions with C-nucleophiles. A central aim of our original proposal was to synthesize and use unsymmetric aryl(auxiliary)iodonium salts in these studies, thereby addressing poor atom-economy of commonly used symmetric salts. This is, however, challenging given the trichotomous reactivity of diaryliodonium salts. The interplay between aryl steric and electronic effects, in what may be considered heteroleptic aryl complexes of hypervalent iodine, and the importance of the counter anion identity is not currently well understood; particularly in respects to their role in dictating a productive reaction pathway (i.e., ionic, radical, aryne) and selective aryl group transfer. Yet, these are critical aspects to consider for future reaction development of unsymmetric aryliodonium reagents and we have uncovered several important features that impact synthesis, stability, and potential synthetic applications of these reagents.
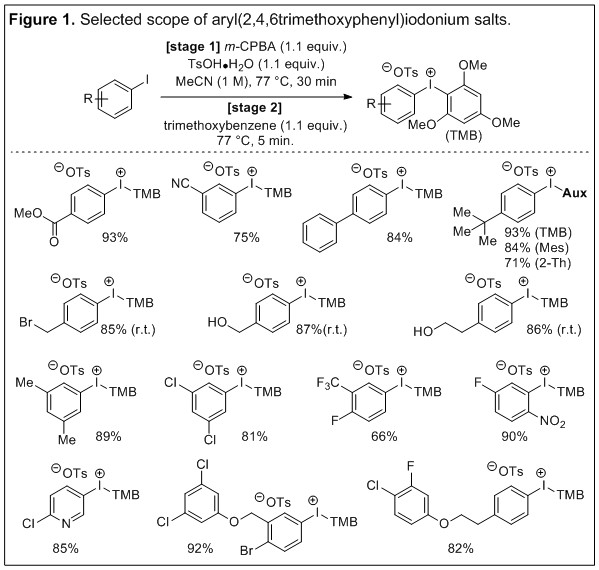
Prompted by reports that suggest the general utility of 1,3,5-trimethoxybenzene-derived auxiliaries and our observations on counter anion stabilization of these salts, we initially targeted aryl(2,4,6-trimethoxyphenyl)iodonium tosylates for study. However, synthetic methods to prepare these salts are underdeveloped and we set out to optimize a practical and broadly applicable “one-pot” synthesis from aryl iodides. Chemometrics was used to monitor and assess the large number of variables associated with condensing multiple steps into a single reaction flask. The variables that are most influential on the yield are aryl iodide concentration (1 M), reaction temperature (77 °C), and the time of stage 1 (30 minutes). The statistically powered optimization of the “one-pot” reaction was carried out efficiently (total of 46 experiments) and does not require chromatographic purification as the products are obtained by filtration after trituration or crystallization. Selected entries from the scope are presented (Figure 1). Both electron-donating and withdrawing substituents are well tolerated on the aryl iodide substrates. Even sensitive functional groups such as benzyl bromide and unprotected hydroxyl groups are unaffected in the reaction when it is conducted at more mild temperature. Moreover, the importance of developing synthetic methods to access unsymmetric aryliodonium salts is highlighted by the use of polysubstituted and elaborate aryl iodides; the corresponding symmetric salts would be more challenging to prepare and more wasteful in subsequent arylation chemistry. We are currently continuing to explore the scope and synthetic utility of these reagents.
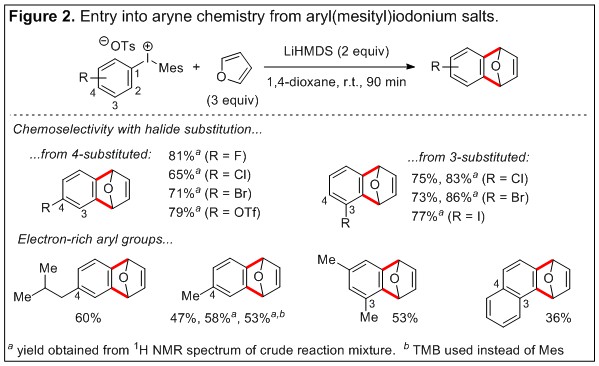
Arynes are versatile intermediates in organic synthesis and the use of diaryliodonium salts as precursors has been known for over 30 years. However, previously developed methods require prefunctionalization of aryl groups with two ortho-activating substituents; the iodonium moiety plays the role of the leaving group in this approach. Alternatively, a C-H deprotonation strategy, that parallels classic methods, circumvents the need for ortho-difunctionalized arenes and constitutes a preparative advantage. However, this pathway has previously been regarded as a low-yielding side reaction that competes with desired nucleophilic addition at the iodonium moiety. We have recently discovered that the C-H deprotonation-induced aryne pathway can become dominant when bulky amide bases (LiHMDS, 1 M in toluene) are used and we have trapped the transient aryne with furan. The heterocyclic cycloadduct that is formed contains two new C-C bonds (Figure 2). Despite being unoptimized this reaction has significant promise for chemoselective late stage functionalization reactions. This is illustrated with the halogenated substrates in Figure 2 and more elaborate substrate in Figure 3. Excellent chemoselectivity is observed for extrusion of the iodonium moiety in the presence of F, Cl, Br, I, OTf-leaving groups at the 3 or 4-position; such high chemoselectivity for polyhalogentated substrates is not characteristic of classic methods. Notably, excellent regioselectivity for deprotonation at the 2-position vs. the 6-position was observed for 3-substituted substrates (Figure 2), and we currently attribute this to proximity-enhanced acidity of the 2-position from the electron withdrawing halide and iodonium moieties. Currently, electron-deficient aromatic rings lead to good yield of cycloaddition product under our unoptimized conditions; however, iodonium salts bearing electron-rich aromatic rings are a limitation and warrant further study (Figure 2).
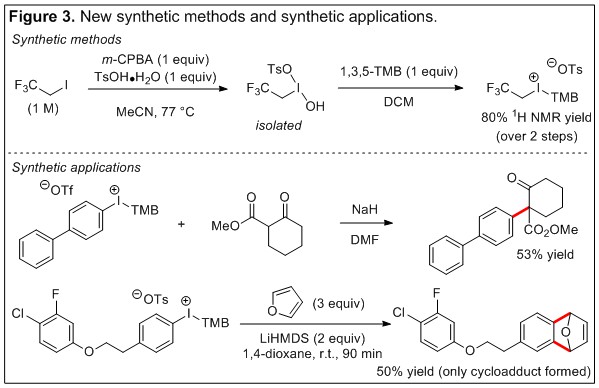
In the next year of the funding period we will continue our efforts in developing new synthetic methods to access unsymmetric aryl(auxiliary)iodonium and fluoroalkyl(auxiliary)iodonium reagents, study their properties and stability, and explore synthetic applications of newly discovered compounds. Progress toward the former has already been achieved (Figure 3, synthetic methods). Synthesis of the Koser’s salt derivative of 1,1,1-trifluoroethyl iodide was performed under the first stage of our “one-pot” method and this was subsequently converted to the 1,1,1-trifluoroethyl(2,4,6-trimethoxyphenyl)iodonium tosylate in good yield (80%, 1H NMR spectroscopy). We are working to transform this result into a practical “one-pot” method. Synthetic applications of aryl(2,4,6-trimethoxyphenyl)iodonium salts already show promise and are demonstrated under unoptimized conditions for electron-rich aryl groups in two representative examples of C-C bond forming reactions (Figure 3, synthetic applications). We are continuing to explore the synthetic applications of unsymmetric aryl(2,4,6-trimethoxyphenyl)iodonium reagents in enolate arylation, aryne-cycloaddition (Figure 3) and other C-C bond forming reactions. We are grateful to the ACS PRF for supporting this research and the educational opportunities afforded to the graduate and undergraduate students who have been involved in it.