Reports: DNI654557-DNI6: Tackling the 'Fire-In-Ice' Problem in the Petroleum Industry: A Molecular Approach
 printer friendly
printer friendlyOur research focuses on studying heterogeneous nucleation of gas hydrates. Hydrates, or fire-in-ice, are crystalline solids comprising guest molecules entrapped in water cages formed through hydrogen bonding between the water molecules. Various gases such as methane, carbon dioxide, and heavier molecules like butane, pentane, and acetone form hydrates. In the current project, we focus on the effects of surface chemistry on the hydrate nucleation process. We will study the nucleation of hydrates near hydrophobic and hydrophilic interfaces using molecular dynamics simulations. To this end we will (i) calculate the nucleation rates near hydrophobic and hydrophilic surfaces (ii) characterize the effect of surfaces on the building blocks of hydrates, i.e. water-mediated interactions between guest molecules and behavior of water cages in SAM-water interfacial regions.
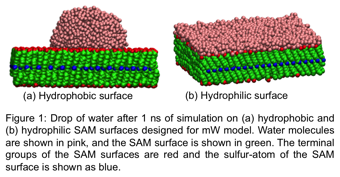
This project report contains the activities from our first year of funding. We have successfully designed hydrophobic and hydrophilic surfaces that are compatible with the water model. We use the monoatomic water model (mW) to perform our simulations. In this model, water is coarse grained and is represented as a single Lennard Jones sphere. The tetrahedrality from the hydrogen bonding between water molecules is imposed as a three-body potential in the Hamiltonian. Because the model does not include electrostatic interactions and hydrogen atoms, it speeds up the simulations by a factor of 5. The ability to sample long simulation times is necessary for studying nucleation. Since mW model is relatively new, limited parameterization to characterize the interactions between different solutes and water have been developed. Therefore, our first step was to develop potentials that can represent hydrophobic and hydrophilic self-assembled monolayer (SAM) surfaces in our simulations. We tested our potentials through simulations of a drop of water on the SAM surfaces. Figure 1 shows the snapshots of a water droplet on these surfaces after 1 ns of simulation. The droplet of water remains a drop on the hydrophobic surface while the droplet spreads and forms a thin film on the hydrophilic surface. This indicates the potentials we developed represent hydrophobic and hydrophilic SAM surfaces.
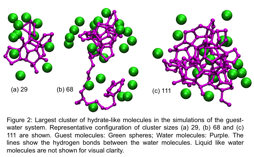
We are currently performing simulations of homogeneous nucleation of sII hydrate. Nucleation is a rare event meaning that the timescales of observing nucleation events are longer than the simulation timescales. To address this challenge we use an advanced sampling technique called forward flux sampling (FFS). In FFS, sampling of the transition from state A to state B is enhanced by dividing the transition into multiple stages and sampling each stage extensively. We have developed software called Scalable FFS (ScaFFS) that enables us to perform large-scale FFS calculations efficiently in high performance computing environment. Using ScaFFS, we are currently simulating homogeneous nucleation of a sII hydrate. Figure 2 shows the initial progress of our calculations where the transition from liquid solution to hydrate is monitored using the largest cluster size of hydrate-like water molecules. The largest cluster size is reported in number of water molecules forming the cluster and hydrate-like water molecules are identified based o their participation in water pentagons. Water pentagons are the basic geometric building blocks in the cages found in the hydrate structures. We have progressed from small hydrate-like clusters to relatively larger clusters and expect that this will continue to grow giving us several trajectories of the transition from liquid-to-hydrate state. This will allow us to quantify the rate of hydrate nucleation and also performed detailed analysis of the molecular mechanisms through which the solution transforms into the solid hydrate-like phase.
We have also studied the lifetime of cages in water as a function of temperature. Cages such as 512 and 51262 form the basic entities of the sI and sII hydrate structure. It has therefore been hypothesized that understanding the behavior, especially the lifetimes of the cages can provide insights into the hydrate nucleation process. To this end, we have started calculations of the lifetime of 512 and 51262 cages in temperatures ranging from 220 to 270 K at pressure of 500 bar. We find that the lifetime of the cages is shorter than that observed in cases using all-atom water models (i.e. models in which water is represented with one oxygen and two hydrogen atoms each with a partial charge). We believe this may be due to the smoothening effect of the Hamiltonian used in mW model. As expected, the lifetime decreases significantly with increasing temperature. Further studies to detail the effects of surface on the lifetime of cages will be performed in the future.
The project involves one graduate and one undergraduate student. This work has provided the graduate student and the PI opportunities to participate in conferences. This has helped in enhancing the professional network and in exposing the students to broad range of research that will be beneficial in their overall growth as a researcher. The involved graduate student has presented at the Gordon Research Conference in August 2015. The PI will present the work at the AIChE National Meeting in November 2015. The undergraduate student has received training in basic concepts of gas hydrates and performing molecular simulations – both of which are not available in usual chemical engineering curriculum. The research experience has helped the student enhance his problem solving abilities, and communication and organizational skills. The results from this work have been used as preliminary results for other grants.










