Reports: UNI154284-UNI1: Alkene Acylation by Photoredox Catalysis
 printer friendly
printer friendlyOverview:
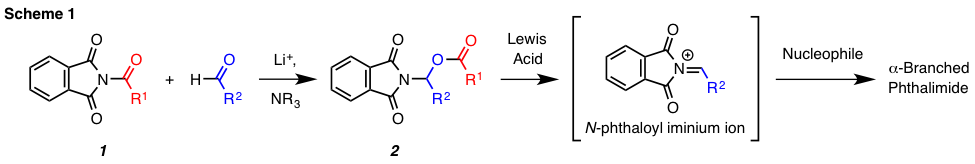
Our group is currently developing reactions involving functional groups that are underutilized in modern synthetic organic chemistry. Specifically, we have found that N-acylphthalimides (cf. 1, Scheme 1) react with aldehydes by a net insertion process to give N-phthalimido-O-acyl-N,O-acetals (cf. 2). This unprecedented process occurs in the presence of lithium cation and a non-nucleophilic tertiary amine. The acyl substituent in the resulting product can be displaced by nucleophilic substitution under acidic conditions to form an alpha-branched phthalimide. This report will describe our initial efforts related to reaction discovery, substrate scope determination, and mechanism elucidation. Additionally, application of this methodology to the acid-mediated synthesis of several alpha-branched phthalimides will be discussed.
Summary of Experimental Efforts: N-Acylphthalimides are an
underutilized functional group in organic chemistry. We were initially
interested in studying the potential for N-acylphthalimides
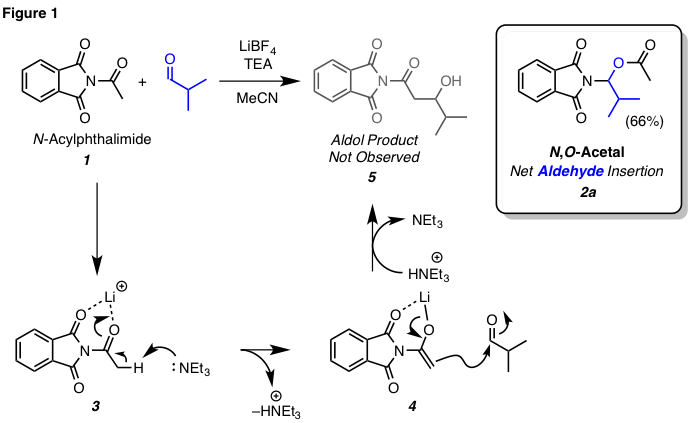
to undergo an aldol reaction. We hypothesized that
lithium cation would coordinate to the imide
carbonyls of an N-acylphthalimide (cf. 3,
Figure 1) and withdraw electron density from the acyl group such that a
tertiary amine base would be capable of deprotonating the alpha-hydrogen atom
(cf. 3 to 4). The resulting lithium enolate
(cf. 4) could form a new carbon-carbon bond via a net aldol reaction. To test this hypothesis, a mixture of isobutyraldehyde and N-acetylphthalimide
(1) was treated with LiBF4 and triethylamine.
To our surprise, the expected aldol adduct 5
was not observed. Instead, N,O-acetal 2a was isolated in 66% yield. Inspecting the
structure of the product suggested that isobutyraldehyde
had undergone a net C–N bond insertion. The exotic nature of the transformation
and the moderate efficiency of the unoptimized
conditions led us to pursue this unprecedented transformation more thoroughly.
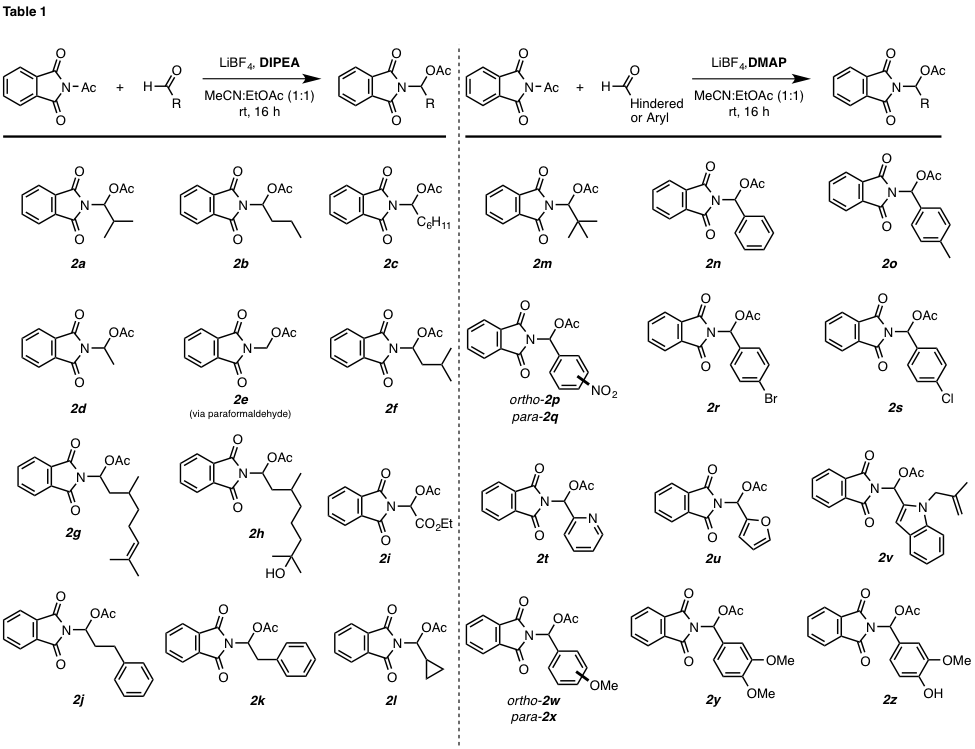
In addition to isobutyraldehyde,
other aldehydes readily participated in the “insertion” reaction to form N,O-acetal products 2b–l
(Table 1). It is noteworthy that the reaction is tolerant of alkenes (cf. 2g),
hindered alcohols (cf. 2h and 2z), reactive
carbocycles (cf. 2l), and aromatic
substituents (cf. 2j, 2k, 2n–z). However, use of benzaldehyde-derived substrates produced the corresponding
phenyl-containing N,O-acetals (e.g., 2n) in low yields. Additionally, no N,O-acetal product was
observed with sterically hindered aliphatic aldehydes
or electron-rich benzaldehydes. This limitation was
overcome when 4-dimethylaminopyridine (DMAP) was used in place of diisopropylethylamine (DIPEA). Under these revised
conditions, pivaldehyde (to give product 2m)
and many aromatic aldehydes (to give products 2n–2z) were found
to participate efficiently under the revised reaction conditions.
Having established that the reaction occurs with a broad
substrate scope, we turned our efforts to elucidating the mechanism of the
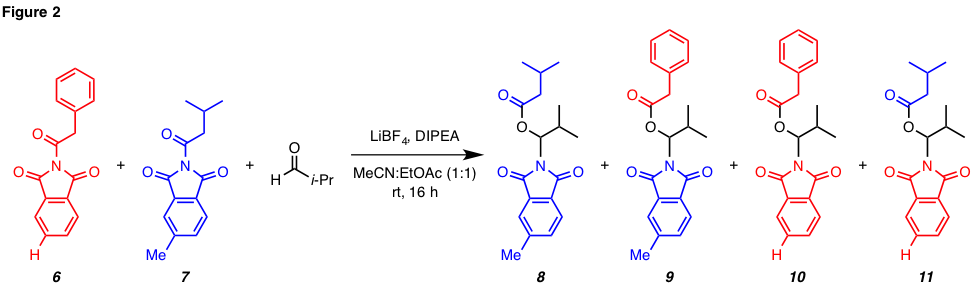
reaction. To determine if the reaction was occurring via a process that
involved acylphthalimide cleavage and phthalimide dissociation, a crossover experiment was
performed by subjecting a mixture of isobutyraldehyde
and N-acylphthalimides 6 and 7 to
conditions that would promote the “insertion” reaction (Figure 2). A
combination of 1H NMR and Hi-Res LC/MS analysis definitively showed
that four N,O-acetals
(i.e., 8–11) were formed and that crossover had occurred. The
crossover observed between 6 and 7 suggests that the acylphthalimide is cleaved under the reaction conditions to
form a reactive intermediate that serves as a strong acylating
agent. This could explain why yields improved when 4Å MS were added to the
reaction mixture because adventitious water was likely engaging the
intermediate acylating agent. Use of N-acetylphthalimide-d3
gave rise to the corresponding N,O-acetal with 30% hydrogen incorporation, which suggests the
formation of a ketene intermediate. However, preliminary ab initio computations
suggest this pathway to be high in energy. Additionally, N-benzoylphthalimide readily participates under the optimal
reaction conditions despite having no alpha-hydrogen atoms.
The synthetic utility of the N-phthalimido-O-acyl-N,O-acetals arising from the
newly discovered transformation has also been evaluated. We have demonstrated
that this class of N,O-acetals is capable of undergoing acid-mediated nucleophilic
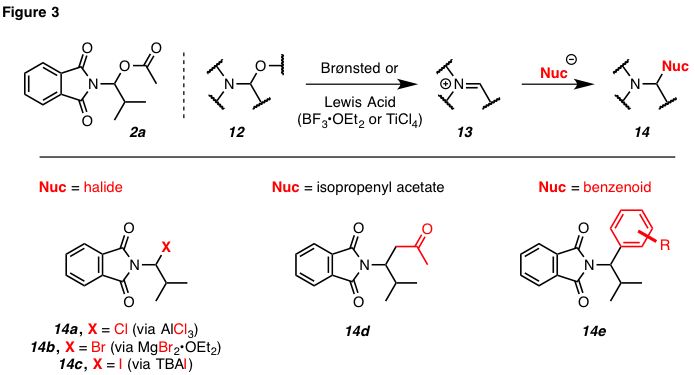
substitution at the acyl substituent (cf. 12–14, Figure 3). For
example, treating N,O-acetal 2a with aluminum trichloride
or titanium tetrachloride gives rise to 14a. Similarly, treating a
mixture of tetrabutylammonium bromide or iodide and N,O-acetal 2a with BF3•OEt2
leads to formation of N,halo-acetals 14b
and 14c, respectively. Carbon-carbon bonds can also be formed via
similar methodology. Specifically, the nucleophilic alkene of isopropenyl acetate readily engages the putative iminium intermediate (cf. 13) to form, after deacylation, ketone 14d. Similarly, nucleophilic benzenoids are capable of reacting with N,O-acetals under Lewis or Brønsted acid conditions to form alpha-branched aryl phthalimides (cf. 14e) in a net Friedel–Crafts
reaction.
In summary, we have discovered an unprecedented method for
preparing N,O-acetals
from N-acylphthalimides. The process involves
insertion of an aldehyde pi-bond into a C–N bond of an acylphthalimide.
The reaction occurs under mild conditions and is applicable to a broad range of
substrates. We are in the final stages of manuscript preparation and will
submit these results for publication shortly. Mechanistic studies are ongoing,
and a fellow Ripon College professor, Joseph Scanlon, has begun evaluating
possible mechanistic pathways using ab initio DFT computations. A total
of nine undergraduate students have made substantial contributions to this
project. In March 2015, aspects of this work were presented at the American
Chemical Society National Meeting in Denver, Colorado via two student posters
and one oral presentation by Patrick Willoughby.