Reports: UR453688-UR4: Transition State Analogues as Mechanistic Probes for Asymmetric Desymmetrization by Cinchona Alkaloid-Based Catalysts
 printer friendly
printer friendlyIntroduction
Enantioselective catalysis by small organic molecules, or organocatalysis, is an important strategy in asymmetric synthesis. However, our understanding of the forces responsible for enantioselectivity in these systems is limited, preventing the more rational design of new catalysts with better selectivity. The goal of this project is to model the intermolecular forces governing enantioselectivity in the asymmetric desymmetrization (ASD) of cyclic anhydrides (Fig. 1). The model we develop can then be used to design or screen potential catalysts, in order to achieve high selectivity with a range of substrates.
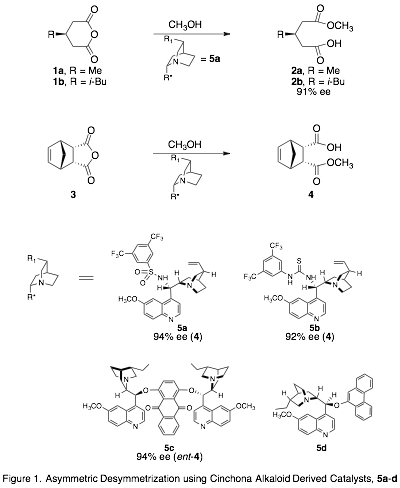
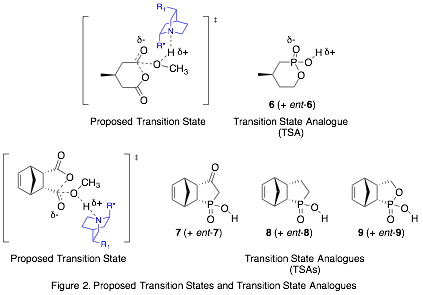
Our approach is to prepare transition state analogues (TSAs) for the organocatalytic ASD reaction and study the interactions between the TSAs and the catalysts, which are derivatives of cinchona alkaloids. The mechanism of this reaction is widely recognized to be a general-base catalyzed process; the quinuclidine nitrogen is thought to activate the methanol by deprotonating it as it attacks the carbonyl center in the rate-limiting step. We are using tetrahedral phosphonic or phosphinic acids as TSAs to mimic the transition state (Fig. 2) and hydrogen bond to the catalyst (Fig. 3). To evaluate our model, we will measure the relative binding affinities of the two TSA enantiomers with catalyst, and correlate that ratio with the enantiomeric excess (ee) of the ASD reaction.
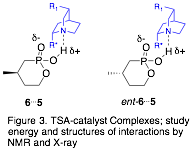
Goals
1. Synthesize TSAs and resolve into pure enantiomers.
2. Evaluate catalyst-TSA interactions by NMR spectroscopy and X-ray crystallography.
3. Measure binding constants for catalyst-TSA complexes and compare these with ee values.
1. Synthesis of TSAs
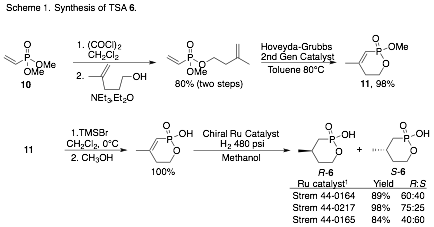
TSA 6:
Both R and S enantiomers have been prepared in 90% ee
or greater (Scheme 1). Ring-closing metathesis, followed by selective deprotection of the methyl ester, 11, and asymmetric hydrogenation
TSA 7:
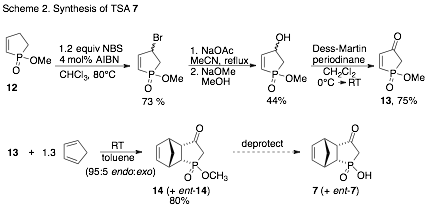
The synthesis of TSA 7 involves a Diels-Alder reaction with dienophile 13 and cyclopentadiene (Scheme 2). Although 13 is a known compound, reportedly prepared in one step by chromic acid oxidation of 12, in our hands the reaction conditions resulted in poor conversions and at most 5% isolated yield. We turned to a more lengthy but reliable route to 13, as outlined in Scheme 2. We are currently in the process of deprotecting ester 14 to obtain 7, and scaling up the synthesis.
TSA 8:
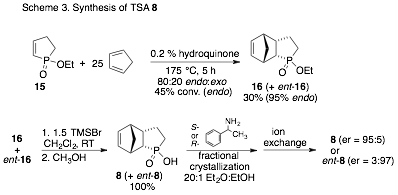
We envisioned that TSA 8 could be prepared via a Diels-Alder reaction between cyclopentadiene and the relatively unactivated dienophile 15. We were gratified to discover that the reaction takes place at 175 °C, providing a 30% yield of the endo adduct. Attempts to improve conversions with Lewis acid catalysts were unsuccessful. After deprotection of 16 with TMSBr, enantiomer resolution was accomplished by multiple recrystallizations with R- or S-phenylethylamine. The free acid, 8, is recovered by ion exchange and we now have sufficient quantities of each enantiomer to pursue NMR studies of the interactions with catalysts.
TSA 9:
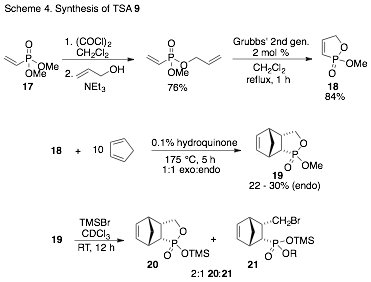
The approach to TSA 9 involves a Diels-Alder reaction with dienophile 18, which is prepared by ring-closing metathesis (Scheme 4). The reaction of 18 with excess cyclopentadiene at 175 °C gives a 1:1 mixture of endo:exo isomers, from which pure endo product can be isolated in about 25% yield. Attempts to use Lewis acid catalysts to effect greater conversions were unsuccessful. Deprotection of ester 19 using TMSBr results in ring-opening to yield 21, in addition to the desired product 20 (Scheme 4). We are currently exploring alternative methods for the deprotection of 19.
Future plans:
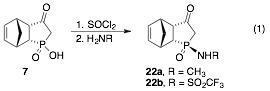
In addition to completing these syntheses, we will resolve enantiomers of TSAs 7 and 9 and assign the configuration of TSAs 7-9 by X-ray crystallography. Future work will also focus on converting the phosphonic or phosphinic acid TSAs to their corresponding amides (eq 1). The amides should provide better mimics of the transition state, because they include substituents that can account for the steric impact of the nucleophile, as well as the tetrahedral nature of the carbon undergoing attack.
2. NMR studies of catalyst-TSA interactions
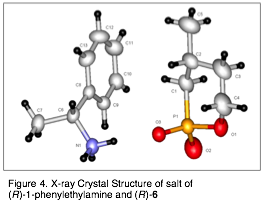
We have begun by studying the interactions of TSA 6 with catalyst 5a. Thus far, we have confirmed the 1:1 binding mode of TSA:catalyst by the method of continuous variations (Job plot). We are also using NOESY NMR to study catalyst-TSA interactions. In addition to pursuing these NMR studies, we plan to crystallize each TSA enantiomer with catalysts, such as 5a, in order to obtain X-ray structures of these interactions.
3. Association (binding) constant measurements
In order to measure binding constants, we have begun NMR titrations using DOSY NMR to obtain diffusion coefficients of the catalyst-TSA complexes at different concentrations of TSA. Currently, we are striving to obtain reproducible results with DOSY.
Impact on Students
Four students have done summer research supported by the ACS-PRF award. They have gained excellent experience in organic synthesis, enantioselective synthesis and chiral resolution, and multi-nuclear and two-dimensional NMR. All of these students are expected to give a formal talk to the chemistry department, give a poster presentation at Carleton or at a national ACS meeting, and write a formal report. All students continue research during the academic year. One of the two Carleton graduates is enrolled in a PhD program in inorganic chemistry at Northwestern University.
Impact on PI
The PRF award was excellent leverage for my successful NSF-MRI proposal (with two co-PIs) for a new NMR spectrometer. A 400-MHz Bruker Avance III NMR system was installed in December 2014 and it has brought advanced capabilities for two-dimensional and DOSY NMR to this project. I attended the Bruker course on DOSY NMR in May 2015 (supported by the NSF-MRI) and I taught my research students to perform this experiment at Carleton.
[1]. Strem 44-0164 is diacetato{(R)-(+)-2,2'-bis[di(3,5-xylyl)phosphino]-1,1'-binaphthyl}ruthenium(II) or Ru(OAc)2[(R)-xylbinap]; Strem 44-065 is diacetato{(S)-(-)-2,2'-bis[di(3,5-xylyl)phosphino]-1,1'-binaphthyl}ruthenium(II) or Ru(OAc)2[(S)-xylbinap]; Strem 44-0217 is Chloro{(R)-(+)-2,2'-bis[di(3,5-xylyl)phosphino]-1,1'-binaphthyl}[(2R)-(-)-1-(4-methoxyphenyl)-1-(4-methoxyphenyl-kC)-3-methyl-1,2-butanediamine]ruthenium(II) (R)-RUCYª-XylBINAP.










