Reports: DNI353760-DNI3: Design of Homogeneous Catalysts for Upgrading Alcohols
 printer friendly
printer friendlyThis PRF grant targets mechanistic analyses to rationally optimize a series of transition-metal based complexes that mediate the oxidant-free catalytic dehydrogenation and dehydrogenative coupling of alcohols. During this reporting period, extensive progress has been made in two key areas: we have (1) developed a complete mechanistic description of the alcohol dehydrogenation reaction and (2) worked to extend this chemistry to first-row (Fe) congeners. Progress in both areas is highlighted below.
Alcohol Dehydrogenation Mechanistic Studies:
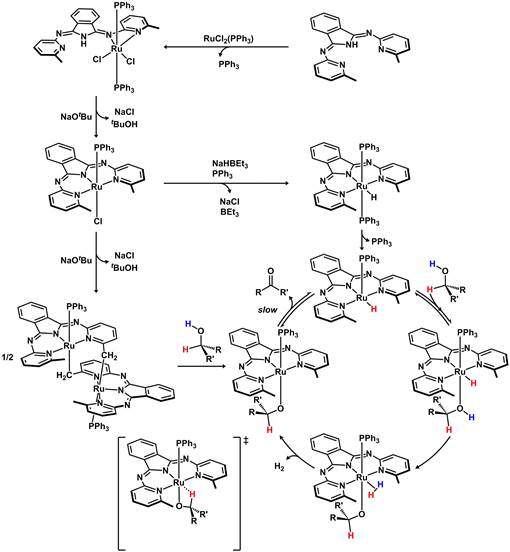
The pincer-based N,N,N dehydrogenation catalyst developed in our research group is among the premier dehydrogenation catalysts available, and promotes base-free and acceptorless dehydrogenation of primary and secondary alcohols, and diols with liberation of dihydrogen under moderate (< 120 In a full paper, we disclosed a detailed mechanistic analysis that included a series of kinetic and isotopic labeling studies, stoichiometric reactions to probe catalytic intermediate species, and new ligand variants to understand the steric and electronic effects of the bis(pyridylimino)isoindolate (bpi) pincer ligand on the dehydrogenation reactivity. Experimental evidence supported an inner-sphere, step-wise pathway for proton and hydride transfers with a β-H elimination turnover-limiting step. Selective isotopic labeling experiments combined with catalyst modification (methylation of the pincer ligand backbone) demonstrated that a cooperative metal-ligand pathway involving the imine functionality is not necessary for efficient dehydrogenation. The activation parameters suggested an associative pathway involving a highly ordered transition state structure. Overall, the data are consistent with a single PPh3 dissociation event from catalyst precursor to generate a coordinatively unsaturated HRu(bMepi)(PPh3) species, which participates in a proton-transfer equilibrium to generate a transient Ru-(H2) alkoxide species. H2 loss affords a ruthenium-alkoxide intermediate that can participate in a turnover-limiting β-H elimination reaction to complete the catalytic cycle (Figure 1). Competition experiments were also examined to elucidate the origin of the chemoselective dehydrogenation of secondary alcohols in the presence of primary alcohols and we found that the dehydrogenation of 1-phenyl ethanol is favored over benzyl alcohol due to slower β-H elimination from the primary alkoxide. To further clarify reaction intermediates, we examined a substrate that cannot undergo β-H elimination (HOCH2CF3) and characterized an intermediate alkoxide (Ru-OCH2CF3). Importantly, we found that the NMR spectroscopic characterization is very similar to the in situ observation an intermediate during catalysis, thus confirming the presence of Ru-OCHR species in the catalytic cycle. Finally, modifications to the pincer ligand revealed that the steric profiles of the methyl groups on bMepi slightly impeded catalytic activity, while electronic modifications of the pincer ligand have a minimal effect on the rate of catalytic dehydrogenation. Overall, these experiments paint a clear picture to support the observed selectivity preferences of secondary over primary alcohols as well as the key contributing factors needed for facile catalysis.
Figure 1. Proposed mechanism of alcohol dehydrogenation and entry into the catalytic cycle from one of several ruthenium N,N,N-pincer based catalysts
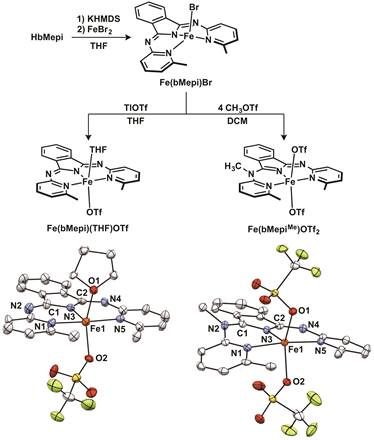
Catalysis with Iron: Based on the success with the ruthenium pincer complexes, and the need for catalysis with base metals, we sought to examine whether related iron variants are similarly active for reductive transformations. We prepared iron complexes with the same pincer-based ligand that was used for the ruthenium dehydrogenation catalysis and demonstrated a late-stage electronic tunability. By modifying a remote imine site on the ligand backbone (by alkylation), the electrophilicity of the metal was adjusted without perturbing the primary coordination sphere. We quantified these changes with electrochemical studies, which confirmed a ca. 400 mV shift in redox couples between the two complexes.
Figure 2. Synthesis of iron-based N,N,N-pincer based catalysts with associated ORTEP structures (50% probability).
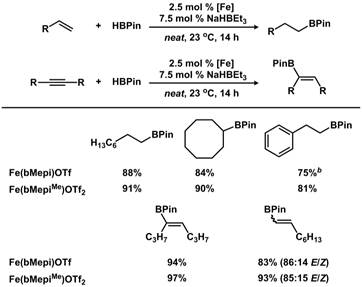
Unfortunately, neither complex was active for catalytic dehydrogenation or hydrogenation reactions. However, both complexes were highly active for a related hydroboration reaction of olefins at room-temperature. Catalysis was achieved under solvent-free conditions, and in addition to their impressive base-metal catalytic activity, a simple post-synthetic modification to the complex enabled improved catalysis and orthogonal selectivity. Using the “modified” iron complex, we found higher activity for the conversion of acyclic and cyclic olefins, styrene, and internal alkynes to the corresponding boronate esters as a single product in all cases, and distinct regioselectivity for internal alkenes, compared with the “unmodified” iron complex. The improved hydroboration activity was quantified by measurement of initial reaction rates, which revealed a four-fold rate enhancement for the “modified” (alkylation at remote site) iron complex when compared to the unmodified variant. Thus, although the target complexes did not behave exactly as anticipated, we discovered a unique class of highly active iron complexes for a related hydroboration reaction.
Figure 3. Scope of catalytic hydroboration of olefins and alkynes by both iron N,N,N-pincer catalysts.
Summary of Support this Period: The support provided by the ACS Petroleum Research Fund has helped to propel the PI’s research in the area of dehydrogenation, and hydroboration catalysis. This is a unique area of the PI’s research efforts that has received considerable attention from the chemical community, as noted by the high citation counts of recent papers. In addition to the PI support, Tim Tseng a graduate student, has been supported by this grant and made considerable progress toward his thesis project. Ongoing efforts during the next reporting period will focus on using the mechanistic results to guide catalyst reoptimization as well as to develop new reactions for alcohol upgrading reactions.