Reports: DNI453824-DNI4: Electrostatically Tethered Catalysts for the Rate Acceleration of H-bond Mediated Transformations
 printer friendly
printer friendly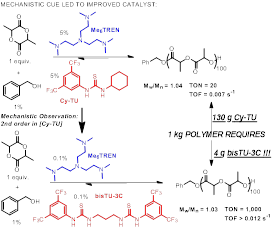

Figure 1. Mechanistic studies in the PI’s laboratory into H-bond mediated ROP have yielded dramatically enhanced activities and productivities without the expense of reaction control.
Narrative Progress Report: The central hypothesis of this proposal was that bimolecular H-bond mediated catalysts systems are artificially slow due to their dispersion in dilute solution; catalysts loadings are typically high (5+ mol%) but usually the reactions are dilute (<50 mM). It was assumed that that tethering the catalysts in solution would increase the reaction rate and offer new mechanistic possibilities. The envisaged coulombic tether, where each cocatalyst would be part of an ion pair, was designed to possess synthetic flexibility yet provide a strong association between the cocatalysts. However, once experimentation began, control studies to identify any preexisting association between the cocatalysts revealed a prominent binding constant between thiourea (H-bond donor) and base (H-bond acceptor) cocatalysts. This discovery launched an un-envisioned area of exploration that has yielded dramatic improvements in the efficiency of these catalysts systems, but resulting from very different structures than those originally proposed.
The first work published as a result of this funding discusses strong binding between H-bonding cocatalysts. Several of the most active H-bonding cocatalyst pairs were shown by 1H NMR binding studies to be highly associated in solution, dominating all other known non-covalent catalyst/reagent interactions during the selected screening reaction (ring-opening polymerization, ROP). The high selectivity and activity which is exhibited by these ROP organocatalysts was attributed to the strong binding between the two cocatalysts, and the predictive utility of these binding parameters was applied for the discovery of a new cocatalyst pair. In a second publication resulting from the grant, the base-dependent reaction mechanism of thiourea alkylamine catalyzed ROP of LA (lactide) was discussed. The binding between cocatalysts was shown to be weaker for the weaker bases, and the mechanism of reaction was shown to be dependent upon the characteristics of the cocatalyst interactions. A mechanistic discovery – of second order behavior in thiourea – during the course of experimentation directly led to a third publication.
Our group translated fundamental investigations into the discovery of the most efficient H-bond mediated catalyst yet known for ROP. This publication describes the rapid rate acceleration of a bisthiourea versus mono-thiourea on thiourea/alkylamine catalyzed ROP, Figure 2. When controlling for the amount of thiourea moiety, the rate of ROP is accelerated regardless of the identity of the alkylamine cocatalyst. Further, the productivity of the ROP is increased with bisthiourea versus monothiourea, Figure 1; this reaction has not been optimized for high productivity. The reaction with bisthiourea was shown to retain excellent control of Mn (by [M]o/[I]o) and narrow Mw/Mn. Preliminary investigation of the effects of bisthiourea upon the ROPs of other ester and carbonate monomers has indicated that rate acceleration occurs regardless of monomer identity, Figure 1. The general high activity and high productivity of bisthioureas as a cocatalysts for ROP suggests a powerful route to advanced H-bonding catalysts. Prior to our disclosure, the most efficient H-bond mediated polymerization required 130 g of thiourea catalyst for the synthesis of 1 kg of polymer (polylactide)! Our new catalyst enables the production of 1 kg polymer with only 4 g of our H-bond donating bisthiourea. The broad applicability of organocatalysts to ROP (i.e. the modes of activation are fundamentally similar across the various monomers and catalysts) suggests that one mechanism informs a diverse set of other catalysts and monomers.
Impact on the career of the PI and research students. The fundamental investigations into the nature of H-bond mediated transformations funded by this grant from the PRF as resulted in the development of the most active H-bond mediated polymerization catalyst known and three peer-reviewed publications in the world’s premier polymerization. The results of this project have facilitated the publication of results in world-class journals and presenting the well-received results of this work at international conferences (Gordon Polymers, June 2015; and ACS Boston, August 2015). The PI has been invited to speak at numerous universities as a result of these presentations and has applied for further external funding to continue these important studies. The research activities funded by this grant have resulted in four student co-authored publications, including a first author publication by an undergraduate researcher. The nature of the studies requires graduate and undergraduate researchers to be trained in a wide variety of chemical methods. In order to successfully complete the proposed research, students were required to conduct catalyst synthesis, polymer synthesis, NMR and materials characterization, thermodynamic studies, chemical kinetics, stereochemical analysis, and air free technique. Fluency in a diverse skillset contributes to a better trained workforce and will facilitate students from the PI’s lab in future employment and research endeavors. The PI has an established interest in working with undergraduates and underrepresented groups in STEM. Two of the three manuscripts published based on this funding have featured undergraduate coauthors including one undergraduate first author and one female undergraduate coauthor. In all, the PI has served as the research advisor for four female (and one male) undergraduate students in his independent career. This funding from the ACS-PRF has laid the groundwork to continue working with undergraduate (and graduate) students, training them in a diverse skillset and producing scientists of the highest caliber.










