Reports: DNI354413-DNI3: Development of Sustainable Co(I) Catalyst Toward C-N Bond Coupling
 printer friendly
printer friendly
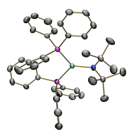
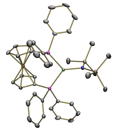
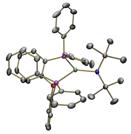
(PPh3)2CoN(SiMe3)2 (dppf)CoN(SiMe3)2 (DPEPhos)CoN(SiMe3)2 µeff = 3.3 µB Co-CNTMS: Bend: 100 oC µeff = 3.0 µB Co-CNTMS: Bend: 100 oC µeff = 2.9 µB Co-CNTMS: Bend: 100 oC