Reports: DNI353298-DNI3: Harnessing Zwitterions for Late-Transition-Metal Catalysis
 printer friendly
printer friendlyThe main thrust of this project over the past year was to begin assessing mechanisms of C–H activation and subsequent/concurrent zwitterion formation (ie, formal positive and negative charge residing at different moieties in the metal complex) using quantum mechanical calculations, specifically Density Functional Theory (DFT) as implemented within Gaussian09. Calculations have been performed on iridium complexes ligated by Cp* and a phosphino-borane ligand of reduced size since structurally and electronically similar complexes have been prepared by the group.
The oxidative addition and zwitterionic isomerization barriers of methane and dihydrogen were calculated by scanning the appropriate bond breaking coordinate (Figure 1). The transition state for the addition step indicates minimal interaction between the substrate atoms and the secondary sphere borane. Consistent with experiment, the addition steps are downhill and favorable. The subsequent zwitterion formation transition state is predicted to be uphill in all cases and only methyl transfer (red coordinate) yields a stable zwitterionic intermediate. Importantly, such a pathway is expected to be accessible energetically at elevated reaction temperatures and pressures.
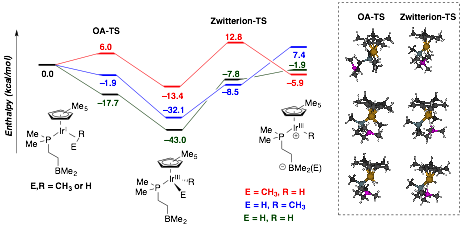
Figure 1. DFT mechanistic analysis of methane and dihydrogen activation at Cp*-Ir(P–B).
Calculations were also performed to force the borane to participate cooperatively in the activation of methane and dihydrogen by blocking one of the two potential binding sites at the iridium center with a ancillary co-ligand (trimethylphosphine, carbon monoxide, or methylcyanide), having varying electronic properties. The identified transition state structures are illustrated in Figure 2 along with the predicted enthalpic transition state barrier. Methane activation is very sensitive to the nature of the ligand with the weak pi-accepting phosphine predicted to be best to facilitate the reaction. The barrier heights are such that increased electron deficiency at the borane coupled with increased electron donation at the metal may drop the barriers to ones manageable at relatively mild temperatures. These predictions await future testing by computation and experiment. Also, interestingly, dihydrogen activation is only minimally affected by the electronic properties of the co-ligand. More detailed analysis of these results should yield important insights into the electronic/structural differences of methane vs. dihydrogen activation.
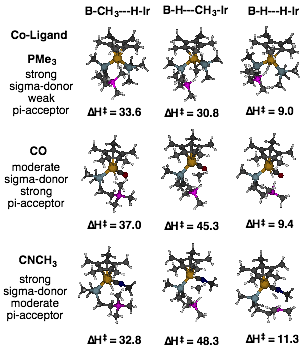
Figure 2. DFT mechanistic analysis of CH4 and H2 activation at Cp*-Ir(P–B)(L) where L = co-ligand.
My research group has also used PRF funding to begin exploring reductive functionalization reactions using catalysts composed of base metals and pincer ligands. We have specifically focused on the bisphosphinite-pyridine (PONOP) pincer ligand as we thought this ligand would have properties that were intermediate between pyridine-diimine (PDI) and bisphosphine-pyridine (PNP) pincer ligated complexes. Gratifyling we identified the PONOP ligand as promoting the regioselective hydromagnesiation of styrene with high yields for Fe, Co, and Ni complexes. Cobalt yielded the best results, indicating the first example of a cobalt catalyst for this reaction (Table 1). The scope of alkenes amenable to this reaction is limited to only electron-rich unsubstituted vinyl arenes at this time or electron-deficient vinyl arenes with an ortho-chelating group, likely involved in the stabilization of the Grignard product (Chart 1). Studies of the mechanism suggest that this transformation is indeed catalyzed by homogeneous PONOP-cobalt and not nanoparticles. Using in situ IR spectroscopy we have begun to study the reaction time course. Interestingly, it appears that at least two kinetic regimes exist depending on the loading of catalyst (Figure 3). The mechanistic information expected from this and related reactions is expected to help us and the community better understand this class of important organometallic reactions.
Table 1. PONOP-base metal catalyst screening table for tandem hydromagnesiation-carboxylation.
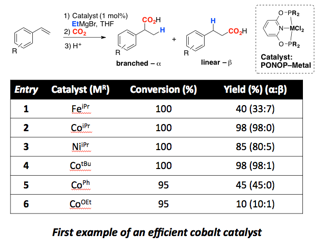
Chart 1. Substrate scope for tandem hydromagnesiation-carboxylation with PONOPiPr-cobalt catalyst.
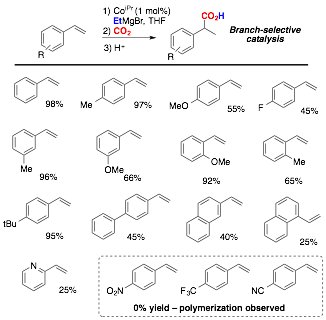
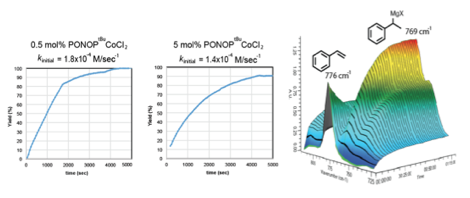
Figure 3. Preliminary React IR data for hydromagnesiation catalyzed by PONOP-cobalt complexes.
In addition to identifying an efficient method to produce benzylic Grignard reagents from electron rich alkenes, we have also very recently discovered that reductive borylation-carboxylation of styrene with a copper catalyst can be achieved under remarkably mild conditions (Figure 4). While such reactions are well developed for alkynes, the result below is the first example of such a reaction at an alkene. Only minimal method optimization has been preformed, so we expect that this reaction will find further improvement in the coming weeks.
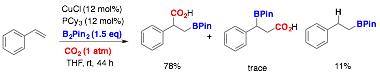
Figure 4. First example of borylcarboxylation of alkenes.
We are also interested in the inorganic coordination chemistry of the PONOP ligand and similar ligands (ie, phosphinite-pyridine, NOP) as these classes of ligand have been only sparingly studied. Visual inspections and subsequent studies of varying phosphine substituents on the PONOP suggest significant structural variation. The alkyl substitutes tBu and iPr lead to trigonal bipyramidal and square pyramidal coordination environments, respectively, based on X-ray diffraction studies (Figure 5). This explains the significant difference in color with the tBu complex being blue while the iPr complex is red. EPR spectra acquired at 115K suggest that only the alkyl substituted PONOP ligands are spin ½ while other PONOP and NOP derivatives have highly broadened spectra, making spin state determinations and other characterization by EPR impossible at this point.
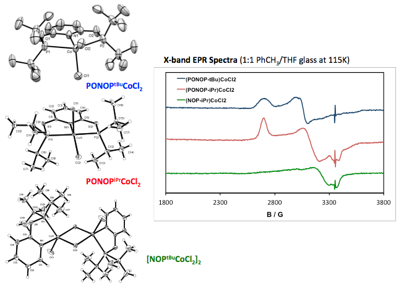
Figure 5. X-ray diffraction structures and EPR spectra for a selection of PONOP and NOP-ligated cobalt complexes.
Finally, owing to the rich phosphine substituent dependent coordination chemistry observed for cobalt, we decided recently to explore nickel and copper coordination chemistry (Figure 6). While the NOPtBu yields expected coordination chemistry for cobalt and nickel, the corresponding NOPPh ligand decomposed in the presence of nickel chloride salt to yield an interesting and rare anionic P(III) pincer ligand (NOPON‑Ph). The coordination environment around copper bromide salts is signifantly impacted by choice of phosphine substritient. Only tBu substituents yield mono-ligated copper coordination chemistry while other substituents (iPr, Ph, OEt) yield coordination complexes with different stoichiometries and geometries.
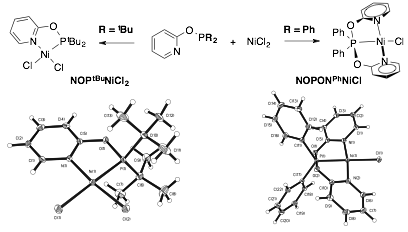
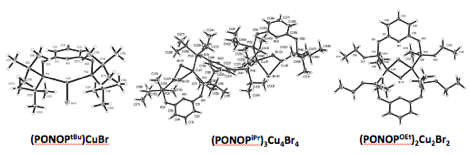
Figure 6. X-ray diffraction structures for a selection of PONOP and NOP-ligated nickel and copper complexes










