Reports: DNI751850-DNI7: N-Alkyl Urea Peptoid Oligomers as a New Type of Self-Assembling, Highly Versatile Soft Materials for Applications Involving Organogelators
 printer friendly
printer friendly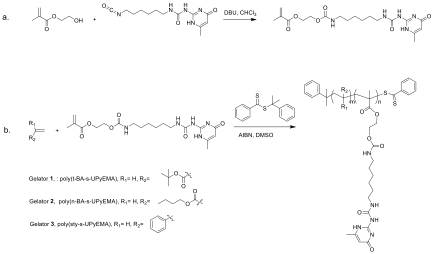
Scheme 1. (a) Synthesis of the UPyEMA monomer. (b) Synthesis of gelators 1-3.
Although the polymers formed stable gels in chloroform we chose to examine the polymer gels in 1,2-dichlorobenzene as it is a higher boiling solvent allowing us to probe the rheology of the gels. The polymers studied in 1,2-dichlorobenenze are described in Table 1. The CGCs are higher in dichlorobenzene compared to chloroform due to the less favorable UPy dimerization.
Table 1. CGC trends for UPy copolymers.
Polymer |
CGC (dichlorobenzene)a |
Monomer Ratiob |
Mn (g/mol)c |
|
Poly(tBA-s-UPyEMA) |
7.8 wt% |
84:16 |
11 900 |
1.45 |
Poly(nBA-s-UPyEMA) |
8.2 wt% |
88:12 |
12 900 |
1.55 |
Poly(Sty-s-UPyEMA) |
6.9 wt% |
86:14 |
12 000 |
1.38 |
aCritical Gel Concentration (CGC)
bMonomer ratio as a percentage
cMn = number average molecular weight
dDispersity
After annealing the gels at 80 °C the storage modulus and loss modulus in 1,2-dichlorobenenze were plotted against time at room temperature (Figure 2).
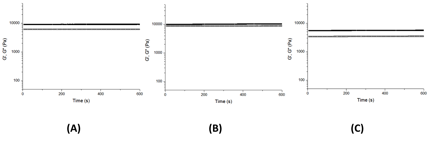
Figure 2. Representative oscillatory time sweeps showing storage modulus (filled symbols) and loss modulus (open symbols) of gels composed of (A) poly(tert-butylacrylate-s-UPyEMA), (B) poly(n-butylacrylate-s-UPyEMA) and (C) poly(styrene-s-UPyEMA).
For all three gels, the moduli remained at equilibrium values with G' higher than G''. While this observation has been used to show the formation of a stable gel,1 compared with most organogels2, 3 our polymers form weak gels with the difference between moduli less than one order of magnitude. Furthermore, the gels in dichlorobenzene appear to be weaker than those formed chloroform. Despite forming weaker gels in dichlorobenzene than in chloroform, we continued to examine the dichlorobenzene gels as interesting behavior was revealed. Figure 3 shows the effect of temperature on moduli. Both G' and G'' decrease with the increasing temperature, with G' decreasing at a faster rate.
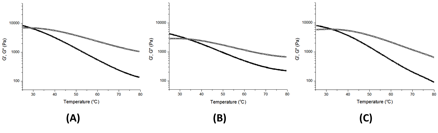
Figure 3. Representative oscillatory temperature sweeps showing storage modulus (filled symbols) and loss modulus (open symbols) of gels composed of (A) poly(tert-butylacrylate-s-UPyEMA), (B) poly(n-butylacrylate-s-UPyEMA) and (C) poly(styrene-s-UPyEMA).
The temperature at which the moduli intersect is the gel transition temperature (Tgel),4 below this temperature the polymer interactions are strong enough to give elastic behavior. These interactions are lost with increasing temperature, resulting in a gel-to-sol transition as the network breaks. The three gels show similar Tgel temperatures around 30 °C. The dimerization constant of UPy is known to be highly temperature and solvent dependent. We speculate that the lifetime of individual hydrogen bonds is decreased under these conditions, leading to the flow observed in the Figure 3. Since the lifetime of hydrogen bonding shortens considerably with increased temperature,5 we believe that above 30 °C the relaxation rate of the polymer chains is faster than the shear rate applied.
Oscillatory frequency sweeps at room temperature were performed on the gels to further study their rheological properties (Figure 4).
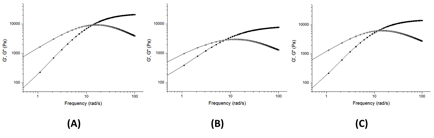
Figure 4. Representative oscillatory frequency sweeps showing storage modulus (filled symbols) and loss modulus (open symbols) of gels composed of (A) poly(tert-butylacrylate-s-UPyEMA), (B) poly(n-butylacrylate-s-UPyEMA) and (C) poly(styrene-s-UPyEMA).
For all gels, the loss modulus is larger than the storage modulus at low frequencies until a crossover point is reached, and the storage modulus becomes larger than the loss modulus. The similar G' and G'' crossover frequency value implies all three gels have the same polymer chain relaxation behavior, which suggests that the gelation is driven by the four-fold hydrogen bonding in UPy. The G' of the tested gels was observed as high as 104 Pa, which is comparable with permanently crosslinked gels.6 Furthermore, the rheology behavior shown in Figure 4 is qualitatively different than that observed for our UPy-functionalized low molecular weight organogelators.1 This is explained by the different gelation mechanisms for the polymer gelators (H-bonding) and low molecular weight gelators (phase separation).
Interestingly, self-healing behavior is typically seen in polymers with rheology similar to that observed in Figure 4.7 Considering the reversible nature of hydrogen bonding, the poly(Sty-s-UPyEMA) gel was subjected to cycles of large amplitude oscillatory strain followed by low amplitude oscillatory strain to examine the time scale of material recovery. Under cyclic deformation at room temperature, the materials shows a clear drop in both moduli values and a gel-sol transition at the onset of high strain (Figure 5). In the transition from high strain to low strain conditions, more than 90 % of the initial mechanical behavior can be recovered in less than 100 s. In other words, our gels are capable of near-immediate recovery following shear thinning.8
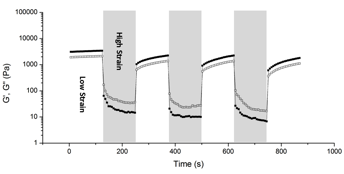
Figure 5. Cyclic deformation of 1 % (unshaded areas) and 200 % (shaded, areas) strain at 20 rad/s on poly(Sty-s-UPyEMA) gel showing storage modulus (filled symbols) and loss modulus (open symbols).
1. X. Chen, P. Fei, K. A. Cavicchi, W. Yang and N. Ayres, Colloid Polym Sci, 2014, 292, 477-484.
2. H. Wang, A. Han, Y. Cai, Y. Xie, H. Zhou, J. Long and Z. Yang, Chem. Commun., 2013, 49, 7448-7450.
3. J. Yuan, X. Fang, L. Zhang, G. Hong, Y. Lin, Q. Zheng, Y. Xu, Y. Ruan, W. Weng and H. Xia, J Mater Chem, 2012, 22, 11515-11522.
4. J. Duan, X. Liang, Y. Cao, S. Wang and L. Zhang, Macromolecules, 2015, 48, 2706-2714.
5. T. Aida, E. Meijer and S. Stupp, Science, 2012, 335, 813-817.
6. H. Shih and C.-C. Lin, Biomacromolecules, 2012, 13, 2003-2012.
7. Z. Wei, J. H. Yang, J. Zhou, F. Xu, M. Zr’nyi, P. H. Dussault, Y. Osada and Y. M. Chen, Chem. Soc. Rev., 2014, 43, 8114-8131.
8. C. B. Rodell, A. L. Kaminski and J. A. Burdick, Biomacromolecules, 2013, 14, 4125-4134.










