Reports: UNI353605-UNI3: Synthesis of Cationic Nickel(II) Complexes Containing Hemilabile Groups for Use as Alkene Hydrogenation Catalysts
 printer friendly
printer friendly
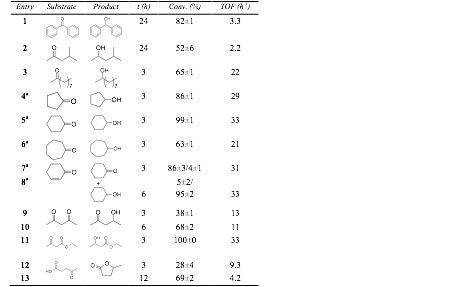
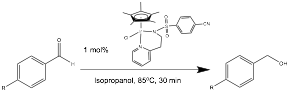
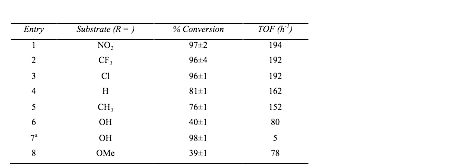
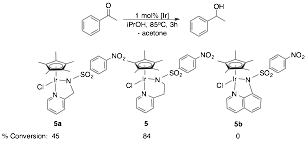
1H NMR spectroscopy used
to determine yield. Yields reported as an average of 3 trials.
1,4-Dimethoxybenzene used as standard. 1.0 M solution of substrate in
isopropanol.
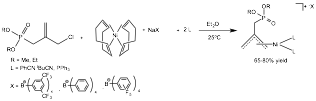
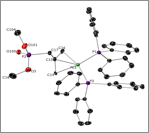
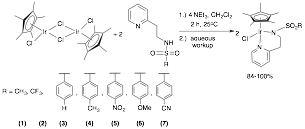
![]()