Reports: DNI654098-DNI6: Transition-State Prediction for High-Throughput Calculation of Accurate Chemical Reaction Rates
 printer friendly
printer friendlyIn brief, we have developed an automated algorithm that predicts transition state geometries, interacts with readily available computational chemistry software to optimize and validate the transition state, then interfaces with an open-source software package to perform transition state theory calculations.
Motivation
Detailed kinetic modeling of combustion has made great progress in recent decades, with models now able to predict and explain complex combustion phenomena for a range of fuels at varied conditions. These models contain many thousands of reaction rate expressions, the vast majority of which are currently estimated. Transition State Theory (TST), coupled with modern computational quantum chemistry methods, would allow such reaction rates to be calculated with high accuracy. As these methods improve and high-performance computers get more powerful, the logical progression is to calculate ab initio all the reaction rates that are currently estimated, or at least the ones to which the model predictions are sensitive. The bottleneck is the human input currently required to guess the geometry of the transition state (TS) – the positions of the atoms at the midpoint of the chemical reaction – required to start a TS calculation. This project aims to predict these TS geometries algorithmically, so that the entire TST calculation can be performed automatically, allowing high-throughput calculation of these important reaction rates.
Progress
During a reaction, most of the reacting molecule closely resembles the geometry of the reactant, which can be predicted using existing distance geometry techniques; the unknown segment of the geometry at the transition state (TS) is at the reaction center, where bonds are being broken or formed. By predicting the distances between a handful of atoms at the reaction center, we are thus able to predict the geometry of the entire TS. We developed a group contribution method to predict the interatomic distances at the reaction center, based on the molecular functional groups reacting. The values for a group are calculated by linear least squares regression on a training set of distances from optimized and validated transition states. The values are organized in a hierarchical tree database, with the top nodes representing the most general template for the reaction. As the tree is descended, the functional groups become more specific, with the most specific groups residing at the base of the tree. If a specific group has not been trained, the group estimation will use the parent group, climbing the tree until a value is found. With a properly designed group tree structure, this allows good estimation of transition states even when training data are sparse.
Using the interatomic distances predicted by our group additive scheme, with distance-geometry methods in the open-source chemoinformatics toolkit RDKit, and constrained optimization with force fields and density functional theory, we have developed an algorithm to create 3D geometry estimates that can be used to start TS optimization searches with quantum chemistry packages.
Once optimized to a saddle point on the potential energy landscape, an intrinsic reaction coordinate (IRC) calculation is performed to verify that the transition state connects the expected reactants and products. This completes the fully automated pipeline that estimates, optimizes, and validates transition states (Figure 1). The optimized and validated transition states are then added to the training data and used to improve the group additive predictions. This self-improving machine-learning aspect of the algorithm is demonstrated in Figure 2, where the predicted distances improve as the size of the training set increases.
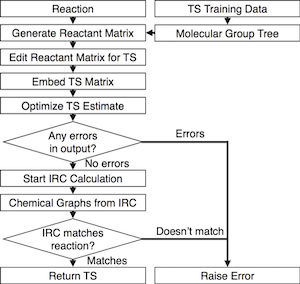
Figure 1. Flow chart for algorithm to automatically generate, optimize, and validate a transition state. From Bhoorasingh and West, Phys. Chem. Chem. Phys. (2015) doi:10.1039/C5CP04706D.
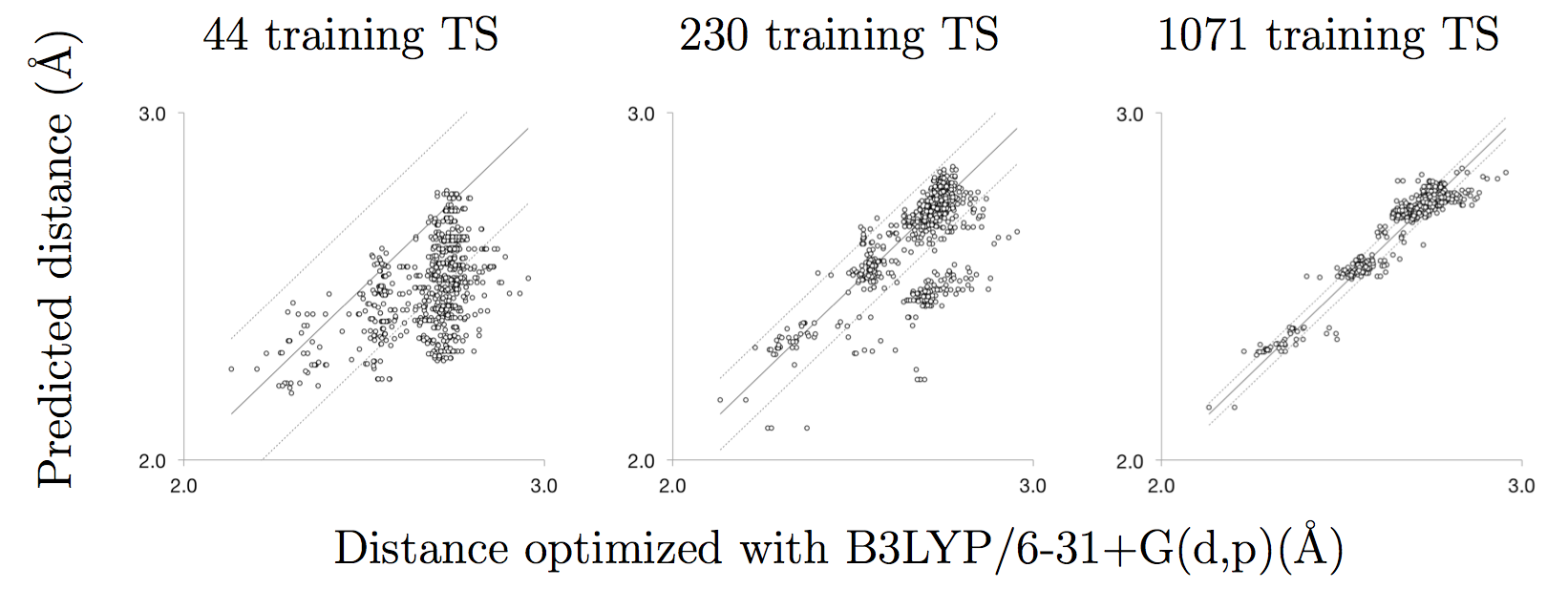
Figure 2: Distances predicted by group-additivity compared to 907 validated hydrogen abstraction transition states found at B3LYP/6-31+G(d,p). Solid line: parity; dashed lines: root mean squared error. Predictions improve as training set size increases. Based on Bhoorasingh and West, Phys. Chem. Chem. Phys. (2015) doi:10.1039/C5CP04706D
We tested the method on a large kinetic model for the combustion of di-isopropyl ketone (DIPK) from the literature. After 2 iterations of re-training the group additive values, we found transition states for 907 of the 1393 hydrogen abstraction reactions in the model, without any human input, and the root mean squared error in the predicted distances for the successful calculations was below 0.05 Å.
Impacts
This project will enable high-throughput TST calculations to provide improved reaction kinetics for automatically generated detailed kinetic models of combustion and fuel processing. Coupled with a separate NSF-funded project to interpret and resolve discrepancies in published kinetic models, the project will quickly impact the combustion modeling community. It will also facilitate several other research projects being undertaken in our research group at Northeastern: predicting the effects of solvents on reaction kinetics, developing reaction rate rules for new reaction families in RMG, and adding reaction kinetics for species containing new elements such as silicon and chlorine. The ACS PRF funding has enabled the graduate student working on this project to present at three major conferences, which has led to interest from industry and academic labs.










