Reports: DNI353254-DNI3: Electronically Responsive Ligands: Toward Two-Electron Reactivity with Earth-Abundant Transition Metals
 printer friendly
printer friendlySelective hydrocarbon oxidation is critical to realizing an energy-efficient chemical industry. Conversion of methane to the 65 billion kg of methanol produced annually occurs through the energy-intensive route shown in Figure 1.[1] Reaching the "holy grail" of replacing this route with direct aerobic methane oxidation has been thwarted by the difficulty of achieving selectivity against mineralization. This problem has been overcome in biology by methane monooxygenases (sMMO and pMMO), where reactivity is achieved by a Cu2O2 core in pMMO and selectivity is controlled by the enzyme architecture.[2] However, the mechanism of methane oxidation by pMMO remains unknown. Support from the PRF has enabled us to target a detailed understanding of methane oxidation by pMMO using uniquely relevant model complexes developed in our group.
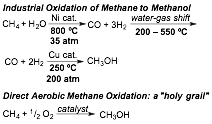
Figure 1. Current route to CH3OH (top) compared to ideal but unrealized direct selective oxidation (bottom).
Oxidatively robust ligands were needed for the study reactive cores relevant to pMMO. Enzymatic studies have revealed that one-electron reduction of the [Cu2O2]2+ core in pMMO leads to the unknown methane-oxidizing core.[2] This presents a challenge to developing model complexes because the comparatively tame [Cu2O2]2+ species are highly reactive. PMMO-relevant [Cu2O2]2+ cores, namely μ-η2:η2-peroxodicopper(II) (P) and bis-μ-oxodicopper(III) (O), have been studied for decades in model complexes; however, supporting ligands in these systems are susceptible to intramolecular oxidation by these cores even at cryogenic temperatures.[3] Therefore, ligands that are oxidatively robust to such cores would enable exploration of more reactive [Cu2O2]+ cores of direct relevance to pMMO. We have developed tBu3tacn, a ligand with unprecedented stability against oxidation.[4] tBu3tacn and other ligands we are developing form the basis for the study of reactive [Cu2O2]+ species that will both inform on the pMMO oxidation mechanism and enable rational design of selective-oxidation catalysts. Furthermore, these ligands serve as platforms for the development of supramolecular assemblies capable of housing reactive Cu2O2 cores within a non-polar cavity, providing a separation of factors controlling reactivity and selectivity. These approaches synergistically allow us to rationally and separately develop an understanding of the two biggest challenges in selective alkane oxidation: achieving reactivity for converting oxidatively resistant alkanes while achieving selectivity for oxidatively susceptible products.
Using PRF funds, we have shown that tBu3tacn is capable of supporting the most stable [Cu2O2]2+ species to date, a P species (1, Figure 2) with a room-temperature half life of 9.6 days.[5] This stability prompted us to explore oxidation catalysis (Table 1). Because of the unprecedented stability of 1, we have explored its electrochemistry, finding the first example of a [Cu2O2]2+ species with a reversible one-electron reduction event (Figure 2), establishing the first direct connection between a model complex and the reactive core of pMMO.
Table 1. Scope of aerobic oxidation by 2.
|
substrate |
product |
catalyst loading (mol%) |
temp. (¼C) |
yield/time |
|
|
|
21 23 19 |
r.t. 50 65 |
91(24h) 73%/4h 70%/2h |
|
|
|
20 5 |
r.t. 50 |
92%/4h 93%/29h |
|
|
|
100 100 100 |
r.t. 50 65 |
0% 9%/76h 34%/76h |
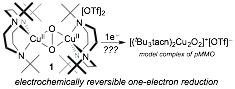
Figure 2. Access to the first pMMO-relevant model complex enabled by tBu3tacn.
The reversible electrochemistry of 1 suggests that reduction may provide an immediate precursor to the methane-reactive species in pMMO, namely CuI/CuII(μ-O2). Such a species is computationally implicated as being in equilibrium in pMMO with its methane-reactive isomer,[6] a CuII/CuIII bis-μ-oxo species. Because 1 is sterically unable to access dicopper bis-μ-oxo forms, we are positioned to be the first to study [Cu2O2]+ species. Furthermore, we were the first to synthesis tBu2Htacn, which is also oxidatively robust but sterically smaller than tBu3tacn (Figure 3). Oxygenation of [(tBu2Htacn)CuI(MeCN)][PF6] generates an equilibrium mixture of 2-O and 2-P, highlighting that relieved sterics allows access to O species. 2 is less stable than 1, likely owing to both the steric accessibility of the Cu2O2 core and access to the reactive O isomer. Importantly, access to O with tBu2Htacn may enable access to the putative methane-reactive CuII/CuIII-bis-μ-oxo core. Our goals are to establish the electrochemical behavior of 2 and to explore its reduction. Therefore, we are positioned to be the first to explore the computationally predicted methane-oxidizing CuII/CuIII-bis-μ-oxo core from pMMO. We will then pursue the synthesis of supramolecular ligand architectures capable of providing selectivity from highly reactive Cu2O2 cores, an example target being shown in Figure 4.
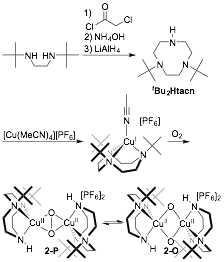
Figure 3. Synthesis of tBu2Htacn and the aerobic copper reactivity it supports.
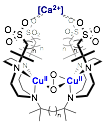
Figure 4. Target for selective oxidation catalysis.
The PRF has been critical to the PI's career, providing funds to advance early research to being fundable by a major funding source with potential for renewal. The PRF-supported research has led to proposals submitted to the NSF and DOE. The PRF funds have also been used to support the PI in presenting the research described above at both of the ACS National Meetings in the last academic year. Furthermore, PRF funding has supported students being trained in challenging organic and inorganic synthesis, in selective oxidation catalysis, and in low-temperature spectroscopic methods. In short, the PRF has enabled the Scarborough group to pursue "holy grail" challenges in selective oxidation catalysis.
[1] Plotkin et al. in Industrial Organic Chemicals, 3 ed., John Wiley & Sons, Inc., Hoboken, NJ, 2013.
[2] Sirajuddin & Rosenzweig, Biochem. 2015, 54, 2283-2294.
[3] Lewis & Tolman, Chem. Rev. 2004, 104, 1047-1076.
[4] Scarborough et al. Inorg. Chem. 2013, 52, 13282-13287.
[5] Scarborough et al. Submitted 2015.
[6] Shiota & Yoshizawa, Inorg. Chem. 2009, 48, 838-845.










