Reports: ND153705-ND1: Organocatalytic Carbonyl-Olefin Metathesis and Olefin Metathesis
Tristan H. Lambert, PhD, Columbia University
The
goal of this ACS-PRF funded project is to develop novel strategies to achieve
double bond metathesis. We have
developed a strategy for catalytic carbonyl-olefin metathesis using simple
hydrazine catalysts. This strategy
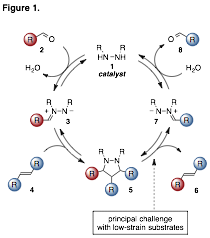
relied on a [3+2] cycloaddition / cycloreversion
strategy, which thus represented a new paradigm for double bond metathesis
(Figure 1). Our first
implementation of this strategy involved the metathesis of aryl aldehydes and cyclopropenes.
The cyclopropenes were necessary because their
high ring strain provided substantial acceleration of both the cycloaddition and especially the cycloreversion
steps. Indeed, for cyclopropenes, cycloaddition was
found to be the rate-determining step, but for most other olefin substrates, cycloreversion was calculated to be rate-limiting,
usually to a very large extent.
We
have been working to identify catalyst modifications that will accelerate the cycloreversion step with less-strained olefin
substrates. In this regard, we have
identified several aspects of catalyst design that we believe may offer
increased rates (Figure 2). Among
these, we believe that angle strain is potentially the most powerful structural
variable. The general idea is to
design catalysts that with larger C–N–N bond angles that will favor
the all sp2-hybridized cycloreversion
products (with preferred angles of ~120¼) while destabilizing the all sp3-hybridized
cycloadducts (with preferred angles of ~109¼).
With
this in mind, we took note that our original catalyst, [2.2.1]-bicyclic
hydrazine 9, had a C–N–N
angle of 104¼, clearly the wrong direction for favoring sp2-hybridization. Notably, we prepared [2.2.2]-bicyclic
hydrazine catalyst 10 with a
calculated C–N–N angle of 111¼ and indeed observed a significant
enhancement in rate for the cycloreversion step with
a norbornene-derived cycloadduct. Encouraged by this result, we have been
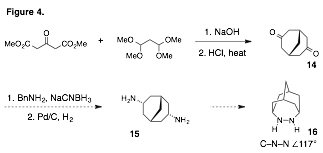
pursuing new hydrazine catalysts with significantly expanded angles. Two such designs include the tricycles 13 and 16 (Figures 3 and 4), which have calculated angles of 116¼ and 117¼
respectively. Our targeted
synthesis of 13 relies on the
intermediacy of the known diketone11, which can be prepared on large
scale from 1,3-acetone dicarboxylate and oxalate
esters. Subsequent reductive amination provides ready access to diamine12, and we are now working to
identify conditions that will forge the key N–N bond. A similar strategy
is expected to furnish the tricycle 16
from the known diketone14 (Figure 4).
Another
catalyst system we are pursuing also possesses large C–N–N bond
angle and, we speculate, may benefit from aromatic stabilization for the cycloreversion step.
Specifically, we are investigating benzocinnoline
derivatives as catalysts for ring-opening metathesis reactions. The general reaction design is shown in
Figure 5, in which benzocinnoline (17) is alkylated by an alkyl halide 18 to produce, after deprotonation, the diazoniumylide19. (This ylide is
identical to azomethine imine 20). Importantly, the diaza-containing central ring is aromatic in intermediate 19 but not in the cycloadduct22. Because of this loss of aromaticity and large C–N–N angle, we speculate
that intermediate 22 will be
especially prone to undergo cycloreversion to the alternative
diazaylide24 and the metathesized olefin 23. Subsequent protonation and Von
Braun dealkylation would then release the new alkyl
halide 25 and return benzocinnoline to the catalytic cycle. Our preliminary
efforts have demonstrated the feasibility of the alkylation equilibrium as well
as ylide formation, and we are currently working to
identify conditions for efficient cycloaddition with
intermediates 19.
In addition to the above efforts, we have also worked on
another approach to carbonyl-olefin metathesis using transition metal
catalysis. To date, a
transition metal-catalyzed carbonyl-olefin metathesis has proven elusive,
mainly owing to the challenges associated with conversion of a metal oxo to the requisite metallooxetane
species.
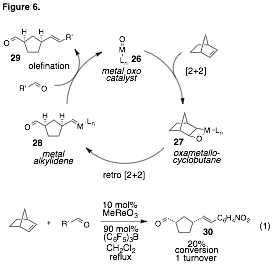
A proposed
Chauvin-type catalytic cycle for a carbonyl-olefin metathesis is shown in
Figure 6. Thus a metal oxo species 26
engages an olefin substrate to form a metallooxetane27. This intermediate undergoes cycloreversion to generate an intermediate 28 bearing a carbonyl and a metal alkylidene. The latter moiety proceeds
to olefinate another carbonyl substrate via metallooxetane formation and cycloreversion,
thereby producing the new olefin 29
and returning the metal oxo to the catalytic cycle. Although the envisioned process is
conceptually straightforward, and many examples of carbonyl olefination
with metal alkylidenes are known, the real challenge
lays in step A: the conversion of the metal oxo
species to the metallooxetane. Nevertheless, evidence
exists that certain metal oxo species can engage
olefins to form metallooxetane intermediates.
Particularly intriguing for our purposes was a recent report from Chen, who
found that certain electron-deficient, four-coordinate
rhenium oxo complexes are capable of catalyzing the ROMP
of norbornene. These results clearly implicate the
generation of a rhenium alkylidene species, which
represents the first half of the catalytic cycle shown in Figure 6 (the
generation of aldehyde functionality was not noted but is presumed). Since it
is established that rhenium alkylidenesolefinate aldehydes, a plausible scenario exists in which
rhenium oxo compounds might serve as viable
carbonyl-olefin metathesis catalysts.
To test this
hypothesis, we treated a mixture of norbornene and nitrobenzaldehyde with 10 mol%
MeReO3 and (C6F5)3B
and observed 20% of the ring-opening carbonyl-olefin metathesis product 30 (eq 1). While this conversion is admittedly
modest, the crucial point is that this result represents turnover of the metathesis
catalyst – a feat that has not been reported in this area. We
subsequently found increasingly electron-rich aldehydes to be significantly
less reactive, and NMR studies reveal that this diminished reactivity is due to
coordination of the Lewis acid by the aldehyde, leaving the Lewis acid
unavailable to activate MeReO3.
Presumably, if the rhenium catalyst could be rendered sufficiently
activated, then Lewis acid activation of the catalyst would not be required,
and the capacity of the rhenium catalyst to engage the olefin substrate would
be dictated simply by the equilibrium of aldehyde coordination by the rhenium.
Toward this end, we propose to synthesize and evaluate a series of
electron-deficient aryl trioxorhenium complexes. Our
studies will initially focus on compounds related to pentafluorophenyltrioxorhenium, which Chen showed to be an optimal
catalyst for norbornene ROMP.
 printer friendly
printer friendly