Reports: DNI654240-DNI6: Mesoscale Simulation and Machine Learning of Asphaltene Aggregation
Andrew L. Ferguson, PhD, University of Illinois at Urbana-Champaign
Synopsis. We have established mesoscale
models of asphaltenes parameterized from all-atom simulations, and used these
models to simulate the self-assembly of hundreds asphaltene molecules at 10 nm
length scales and microsecond time scales. By combining coarse-grained models
parameterized by atomistic data with high performance simulation software
running on GPU hardware, we have performed the first molecular dynamics
simulations to attain such length and time scales with molecular-level detail,
thereby permitting direct simulation of the complete assembly hierarchy posited
by the Yen-Mullins model. The morphological trends observed in our mesoscale
simulations are in broad agreement with Yen-Mullins, in that we observe
hierarchical assembly from monomers to nanoaggregates to clusters to a spanning
viscoelastic network. Providing molecular-level insight of the assembly
mechanisms, we have uncovered new understanding of asphaltene assembly as a
function of concentration, temperature, and chemistry. This work supports the
professional development of the graduate researcher by providing him training
in molecular simulation, statistical mechanics, and scientific writing, and
this year he will attend a national conference (likely APS) to present this
work. This work also permits the PI to leverage his training in molecular
simulation and statistical mechanics to establish himself as an independent
young investigator in a new field with credentials in self-assembly and
mesoscale simulation.
Mesoscale model
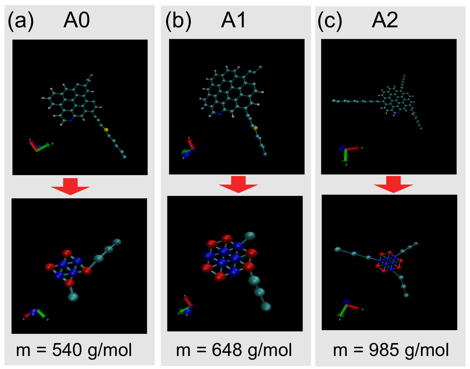
construction. We have constructed mesoscale models of three
prototypical asphaltene molecules – A0, A1, and A2 (Fig. 1) – selected to assess the influence of the size of the
aromatic core and number of aliphatic side chains on assembly. We conducted
all-atom simulations of monomers and dimers of each
architecture in heptane solvent using the GROMACS simulations suite
employing the GROMOS 54a7 force field. We generated coarse-grained mappings of
each molecule under the coarse-grained Martini force field lumping ~4 atoms
into each coarse-grained bead to enable accelerated simulations by both
removing degrees of freedom and smoothing the potential energy landscape. The
distribution of bond lengths, bond angles, torsional angles in each
coarse-grained model were adjusted to match those observed in the all-atom
model simulations by optimizing the baseline Martini bonded parameters using
Boltzmann inversion. The non-bonded interactions in the coarse-grained model were
adjusted such that the potential of mean force (PMF) for dimerization matches
that computed in all-atom calculations (Fig.
2).
Fig. 1 – Prototypical asphaltene molecules
(a) A0, (b) A1, and (c) A2 studied in this work. Each panel shows the all-atom
representation of the asphaltene molecule, its coarse grained mapping under the
Martini model, and its molecular weight.
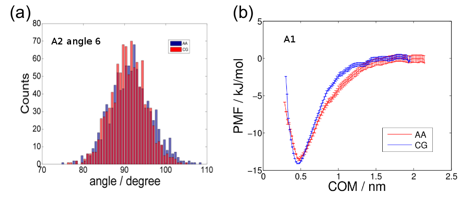
Fig. 2 – The standard Martini
bonded and non-bonded interaction parameters were adjusted to match the
intramolecular distributions of bond lengths, angles, and torsions and the
dimerization PMF observed in all-atom simulations. (a) As an illustration of the
Boltzmann inversion procedure, the Martini force constant for the harmonic
bending of angle #6 in molecule A2 was adjusted by Boltzmann inversion to match
the distribution of bond angles observed in an all-atom simulation. (b) As an
illustration of the PMF matching procedure, the Martini non-bonded parameters
for molecule A1 were optimized such that the dimerization PMF matched that
computed in all-atom calculations.
Mesoscale assembly
mechanisms. Using our coarse-grained model, we have simulated the
assembly of up to 250 asphaltene molecules in heptane solvent over a
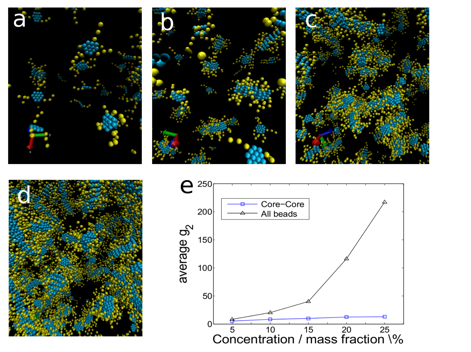
concentration range of 5–25% mass fraction. In the case of A2, we observe
the hierarchical formation of nanoaggregates, clusters, and ultimately a
gelation transition to a viscoelastic network as we elevate concentration (Fig. 3). Nanoaggregates form by
stacking of the aromatic cores possessing a fractal dimension of ~1.1, whereas
clusters of nanoaggregates form by interactions between the asphaltene side
chains, forming a connected network with a fractal dimension of ~2.3. The relative
scaling of the energetic and entropic contributions to nanoaggregation
sets the characteristic number of molecules within a nanoaggregate. The
entropic penalty associated with restricting the configurational entropy of the
aliphatic side chains within a nanoaggregate compared to a free monomer should
be mitigated for asphaltene architectures possessing fewer side chains, leading
to an elevated driving force for assembly. Indeed, simulations of asphaltene
architecture A1 show there to be no characteristic size of a nanoaggregate,
with a downhill thermodynamic driving force for the unbounded kinetic agglomeration
of pseudo-1D stacks.
Fig. 3 – Asphaltene assembly
hierarchy for molecule A2. (a) At concentrations <5% mass fraction we
observe monomers and small oligomers. (b) In the range 5–10% mass
fraction, nanoaggregates with a characteristic cluster size of ~8 molecules
form. (c) For mass fraction range 10–15% we observe hierarchical
aggregation of the nanoaggregates into clusters of nanoaggregates. (d) For mass
fraction >15% we observe a gelation transition to form a viscoelastic
network forming a giant connected component containing all molecules in the
system. (d) The mass-averaged mean cluster size as a function of concentration illustrates the existence of two length scales, hierarchical
assembly, and the gelation transition. Defining two molecules to be within a
connected cluster if any of their core atoms are within a cutoff of rc = 0.55 nm characterizes the mean size of a nanoaggregate
formed by pseudo-1D stacking of the aromatic cores, which is a weak function of
concentration. Defining two molecules to be within the same connected cluster
if any of their core or side chain atoms are within the cutoff characterizes
connectivity within and between nanoaggregates, revealing the formation of
clusters fro mass fractions of 5–15%, and a gelation transition at 15%,
beyond which all asphaltene molecules exist in single giant connected
component.
Future work. We continue to characterize
the morphology and mechanisms of hierarchical assembly and compare to published
experimental measurements. A publication detailing our work to date is in
preparation. We intend to support our mechanistic analyses by performing
machine learning of the aggregation pathways using multi-body nonlinear
dimensionality reduction techniques developed in our lab. We will also study
the thermodynamic stability of the various hierarchical aggregates using
alchemical and expanded ensemble methods, and the kinetics of assembly through
cluster lifetimes and Markov state models.
 printer friendly
printer friendly