Reports: ND153385-ND1: Borenium Catalysts for Metal-Free Enantioselective Reductions and Petasis Reactions
Cathleen M. Crudden, Queen's University
In the first year of
our study we developed Lewis acidic meso-ionic carbene (MIC) borenium ions
based on a 1,2,3-triaza-5-ylidene motif that effectively catalyze hydrogenation
under exceptionally mild conditions (see previous report).1 In year two, we examined C–C bond
forming reactions and enantioselective reductions.
C–C bond forming
reactions. Previously
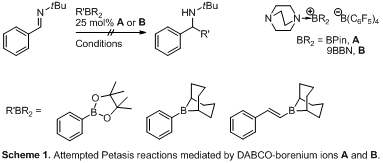
synthesized borenium ions A2 and B were examined under a wide variety of
conditions for the reaction of phenyl borane or vinyl borane with an imine in
an attempt to transfer an alkyl or aryl substituent (Scheme 1). However, at
ambient temperature no reaction was observed and under forcing conditions at
elevated temperature borenium ions A and B lack stability. Since
no trace of product was formed, we focused attention on enantioselective
reduction chemistry.
Enantiopure MICs and enantiopure
boranes. One
of the challenges with the development of enantiomerically pure MICs is the
propensity for deprotonation and concomitant racemization at wing tip groups on
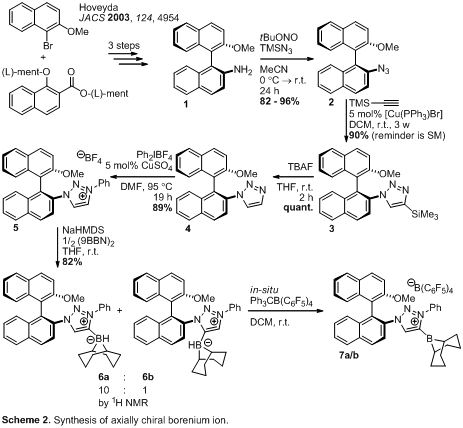
nitrogen (See first report). Thus we focused on axially chiral MIC-borenium
derivatives as shown in Scheme 2. Known amine 13 was converted to azide 2 by careful reaction
with tert-butyl nitride and trimethylsilylazide. The challenging
1,4-selective Huisgen-cycloaddition of the two sterically encumbered substrates
2 and trimethylsilylacetylene required extensive screening that revealed
[Cu(PPh3)Br] as the most competent catalyst, albeit with extended
reaction times. Triazolium 5 was prepared from 3 using Cu-catalyzed
N-arylation after silyl-deprotection.4
Treatment of 5 with base followed by 9BBN gave boranes 6a/6b
in a 10:1 ratio.1 Hydride abstraction
using Ph3C+B(C6F5)4–
gave the borenium ion mixture 7a/b. All reactions up to 5 were
high yielding processes amenable to gram scale synthesis.
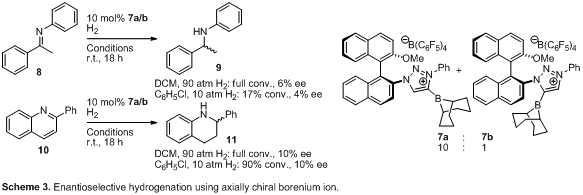
Borenium-ion mixture 7
was tested in hydrogenation reactions despite the inseparable regio-isomeric
mixture (Scheme 3). Low chiral induction for reduction of ketimine 8 and
quinoline derivative 10 was observed. Since NHC–derived
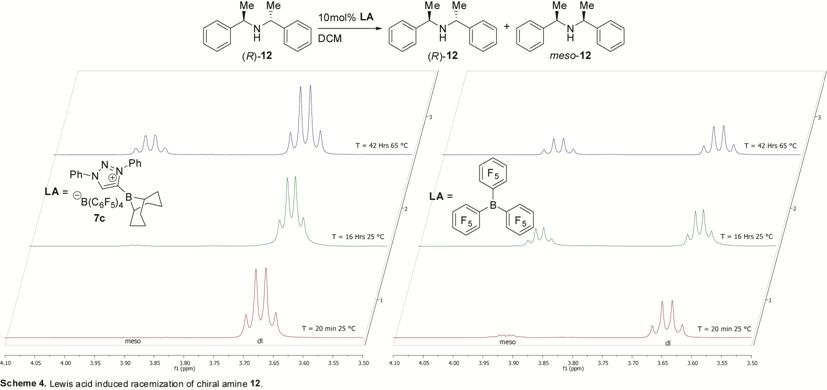
borenium ions are known to racemize enantioenriched amines,5 we exposed enantio-pure amine (R)-12
to catalytic amounts of achiral borenium ion 7c. This gave only traces
of meso-12 by 1H NMR under conditions similar to
catalytic hydrogenations, while B(C6F5)3
showed considerable racemization (Scheme 4). The low degree of enantio
induction is therefore likely attributed to poor differentiation of the two
prochiral faces of the substrate, or to competing selectivities of 7a
and 7b in the hydride delivery step.
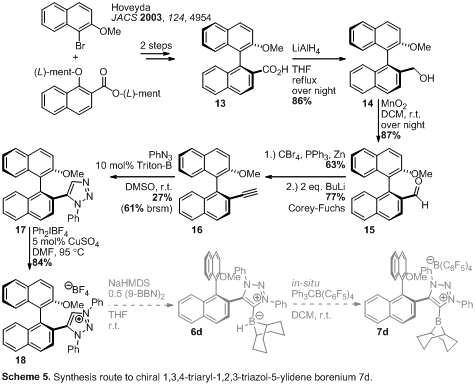
We therefore targeted
MIC-borenium ion 7d, which would give rise to a single isomer (Scheme 5).
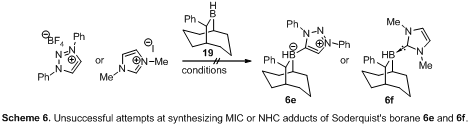
We also chose to prepare 7e and 7f derived from the chiral 9BBD
borane 196
(Scheme 6).Two routes to the
linchpin carboxylic acid 13 were examined: one described by Hoveyda3 and another by Hayashi7 (not shown) that starts with cheap, commercial
BINOL and catalytic carbonylation chemistry to provide 13. Access to the
alkyne 16 via a Sonogashira route in high yield was unsuccessful despite
extensive screening. Alkyne 16 was accessed by a Corey-Fuchs reaction
sequence (reduction to benzylic alcohol 14, oxidation to aldehyde 15,
then Wittig reaction followed by elimination of bromide) and subjected to a
base-catalyzed 1,5-selective Huisgen cyclization8
with phenylazide in moderate yield. The 1,5-diaryl-1,2,3-triazole 17 was
N-arylated to give the 1,3,5-triaryl-1,2,3-triazolium tetrafluoroborate 18.
With 18 in hand, future work will be to synthesize the corresponding
MIC-borenium 7d. We also plan to explore the use of triazolium salts 5
and 18 as ligand precursors in transition metal compounds.
Unfortunately, all
attempts at generating and isolating either adducts 6e or 6f of
Soderquist's borane 19 failed likely due to excessive steric hindrance
of the borane combined with the limited stability of the carbenes under more
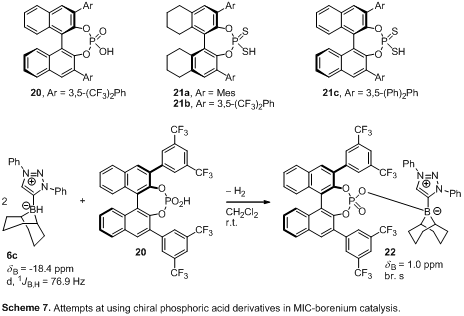
harsh conditions.Chiral anions. Considering the limited
success of both chiral MICs and boranes to support catalytic borenium reduction
chemistry, we focused on a chiral anion strategy based on known phosphates and
thiophosphates.9Exploring a commercial
phosphate and a few synthesized thiophosphates10
(20, 21a-c; Scheme 7) we found only stoichiometric amounts of reduction
products, suggesting that this system fails to activate H2 and
achieve catalytic reduction. Mixing equimolar amounts of MIC-borane 6c and
20 gave rise to the formation of a 4-coordinate boron compound 22
that does not dissociate phosphate sufficiently for H2 activation
even under forcing conditions (heat, elevated pressure). We are currently
synthesizing more sterically hindered phosphates with large groups in the 3,3'
position of the BINOL-backbone to minimize catalyst deactivation by
coordination to the B-centre.
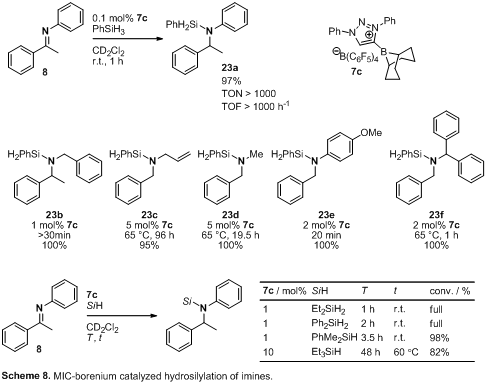
Hydrosilylation. As an alternative to
hydrogenation, we investigated the hydrosilylation of ketones and imines using
MIC-borenium ions and precursor boranes.11 This procedure is simple, and cleavage of H-Si bonds
is known to be easier than H2 activation. We were pleased to find
that 7c efficiently catalyzed the reaction of PhSiH3 and 8
at very low catalyst concentrations, high substrate to catalyst loadings (currently
as high as TON 1000) and promising rates (TOF 1000 h–1) at
ambient temperature (Scheme 8)! Importantly, less sterically hindered
N-substituents such as benzyl, allyl and methyl resulted also in efficient
catalytic hydrosilylation. In addition, a number of silanes are tolerated in
this chemistry.
Unfortunately, when
hydrosilylation reactions with PhSiH3 and 8 or 10 as
substrates were attempted under the same conditions as depicted in Scheme 3,
using the 7a/b mixture as the catalyst, the same poor degree of
induction was observed. However the high reactivity observed for
hydrosilylation will provide us increased opportunities to design effective
catalysts that may have better overall enantioselectivities. This work will
continue past this granting cycle. In addition, we will continue to explore this
exciting silylation chemistry and plan to publish the results in due course.
 printer friendly
printer friendly