Reports: DNI652732-DNI6: Atomic-Scale Visualization of Excitonic States in Individual Polymer Molecules
 printer friendly
printer friendlyPolymer materials owe their success to the enormous amount of research carried out to understand their properties. Despite this large body of knowledge, a fundamental aspect of polymer properties—the relationship between the exact polymer chain structure and electronic properties—remains poorly understood owing to the lack of tools capable of probing this relationship. This work elucidates the impact of structural disorder on the electronic properties of model oligomer molecules by using advanced real-space spectroscopic techniques based on Scanning Tunneling Microscopy (STM). Two types of oligothiophene molecules (DDQT-1 and DDQT-2, Figure 1) were used as finite-length models for investigations of structural effects in polymers. Each oligomer contained a conjugated backbone composed of thiophene rings, with alkyl chains attached to the backbone. DDQT-1, containing four thiophene rings, was used as a useful model for isolating the effects of short-range disorder on the electronic structure, while DDQT-2, containing eight thiophene rings, served as a basic model for long-range disorder. The experiments were performed using a home-built cryogenic ultra-high vacuum (UHV) scanning tunneling microscope (STM). In two sets of experiments, DDQT-1 and DDQT-2 molecules were deposited on an atomically flat Au(111) surface, in situ via sublimation.
Figure 1. Didodecyl-quaterthiophene oligomers DDQT-1 and DDQT-2.
STM imaging of a dilute coverage of DDQT-1 molecules on Au(111) revealed a variety of conformations (Figure 2). DDQT-1 molecules on Au(111) predominantly formed dimers linked by their alkyl substituents and, instead of adopting the trans-conformation found in bulk oligothiophene crystals, assume cis-conformations (Figure 2a), even though trans-conformers were also observed (Figures 2b and c). Surprisingly, in our scanning tunneling spectroscopy (STS) measurements of the electronic structures of DDQT-1, we found that the impact of the conformation was not decisive in determining the LUMO energy (Figure 2i). Instead, we found that the orbital energies vary substantially due to the local variations in the Au(111) surface reactivity.
At higher sub-monolayer molecular concentrations, the molecules self-assembled into ordered islands (Figure 3a). Simultaneously, the predominant molecular conformation converted from cis to trans (Figure 3c), with molecules comprising self-assembled islands showing qualitatively different registry with the Au(111) lattice, as compared to that found at smaller surface concentrations (compare Figures 2e-h to Figure 3d). Further structural transformations were found at still higher molecular concentrations, with molecules showing a different type of the trans conformation and registry with the Au(111) lattice. Electronic structure measurements of molecules in self-assembled islands suggest that the orbital energies and spatial distributions vary substantially due to the variations in local molecular environments, as well as due to the variations in the Au(111) surface reactivity.
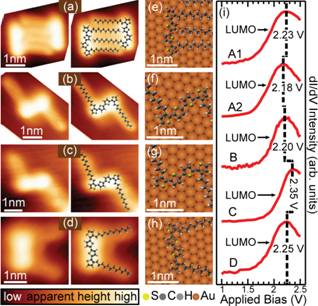
Figure 2. DDQT-1 molecules on Au(111). (a)-(d) STM images DDQT-1 molecules in different cis- and trans- conformations. (e)-(h) Atomic models of DDQT-1 1molecules on the Au(111) surface lattice. (i) STS of the LUMO state for molecules from (a)-(d).
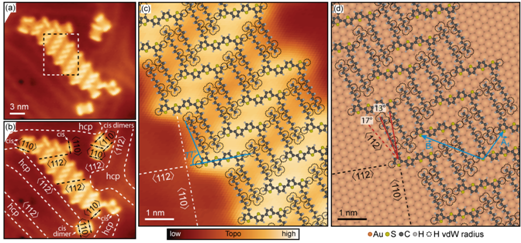
Figure 3. DDQT-1 molecules on Au(111) in the high coverage regime. (a) STM image of a 2D crystals of DDQT-1 molecules. (b) STM image from (a) with indicated molecular orientations and Au(111) crystallographic directions. (c) STM image with overlaid molecular structures of part of the crystal indicated in (a). (d) Atomic models of DDQT-1 backbones on the Au(111) surface.
To investigate the effects of long-range conformational disorder on the oligothiophene electronic structure, we have carried out experiments on the longer oligothiophene, DDQT-2. STM imaging of DDQT-2 on Au(111) showed self-assembled chains of molecules tethered to each other with alkyl chains, analogously to the crystal-like structures observed for DDQT-1. A wide variation of straight and curved conformations was found. In addition, the molecules adopted distinct torsional conformations with alkyl ligands forming cis- or trans- mutual orientations. For each conformer of DDQT-2, by using STS mapping, we identified a progression of particle-in-a-box-like states corresponding to the LUMO, LUMO+1 and LUMO+2 orbitals (representative data for one such conformation is shown in Figure 4). Analysis of orbital energies obtained from STS data revealed very similar molecular electronic structures for different possible molecular conformations. By using density functional theory calculations, we showed that the lack of variation in electronic structures among the different oligothiophene conformers implies that the effect of the Au-oligothiophene interaction is nearly identical for all studied torsional conformations.
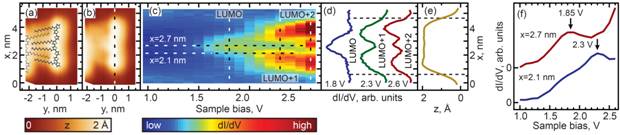
Figure 4. STS mapping across a cis-cis-cis (CCC) conformer of DDQT-2 molecules. (a) STM image with an overlaid atomic model of the molecule. (b) The path of STS mapping (dashed line). (c) DOS as a function of the bias voltage and position x along the path shown in (b). (d) LUMO, LUMO+1, and LUMO+2 DOS along the same path. (e) Backbone profile. (f) STS spectra measured at locations indicated by horizontal lines in (c).
Our results show that a significant degree of structural as well as electronic diversity can be expected even in a relatively simple system composed of short substituted oligothiophenes adsorbed on a metal surface. Diverse self-assembly regimes of DDQT molecules on the Au(111) surface result from competition between the different types of molecule-metal and molecule- molecule interactions, including the surface-thiophene atomic-level interactions and van der Waals interactions involving the alkyl ligands, thiophene backbones, and the Au surface.
The work described above was performed by three graduate students (Christian Gervasi, Dmitry Kislitsyn and Ben Taber) and an assistant professor (George Nazin). A postdoctoral researcher (Yinghao Liu, now at Seagate) constructed an STM system capable of optical spectroscopy and an undergraduate researcher (Matt Robertson, graduated) built the thermal evaporator used to deposit the oligothiophenes. In addition to these members of the team, one graduate student (Jon Mills) and two undergraduate students (Ariel Rosenfield and William Crowley) are now contributing to related research in our lab. In this work, we are aiming to extend our STS approach to longer oligomer molecules on surfaces where the impact of surface-molecule interaction could be reduced, and a more diverse set of conformational variations could be probed. Support from this New Investigator grant was crucial for training of participating students and postdoctoral researcher, who, at the beginning of this project, were starting their work in the lab.










