Reports: DNI653674-DNI6: Quantum Mechanical Calculations of Large-Scale Explicit Solvation
 printer friendly
printer friendly
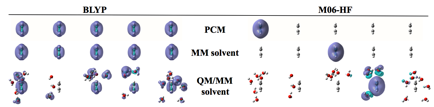
One approach to modeling both the short- and long-range
effects of solvation is to partition the system into a quantum mechanical
region (QM) and a classical region treated by molecular mechanics (MM) or a PCM.
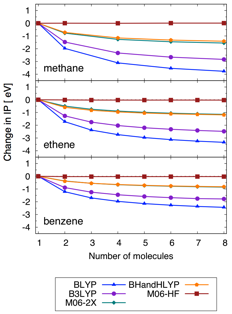
Ideally one would like to include enough solvent molecules in the QM region to
yield a converged result. Our results show that the amount of solvent required in
the QM region to compute converged spectra depends on the basis set (see Figure
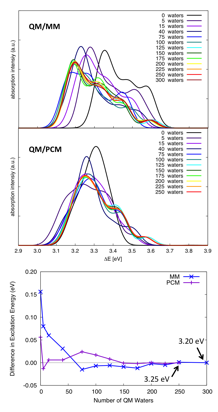
3) and the character of the excitations, rather than the charge on the solute. The QM/PCM model requires fewer QM solvent molecules than
QM/MM to reach convergence (Figure 4). The QM/MM and QM/PCM models converge to
different excitation energies for the main peak (~0.05 eV difference) for our
tests with anion solutes, and the QM/PCM model leads to a significantly more
intense HOMO to LUMO peak compared to the QM/MM model. When comparing QM/MM and QM/PCM for ionization,
non-equilibrium PCM must be used to take into account the vertical ionization
that is modeled when no relaxation of the solvent positions are considered. In
this case PCM again shows accelerated convergence compared to an MM point
charge water model, requiring 25-50 explicit solvent molecules (one solvation
shell) to reach a close to converged value.