Reports: DNI453861-DNI4: Understanding the Molecular Mechanism of Biological Desulfurization to Improve Sulfur Removal from Petroleum
 printer friendly
printer friendlyWe have begun elucidating the catalytic mechanisms of 2'-hydroxybiphenyl-2-sulfinic acid desulfinase (DszB), an enzyme responsible for cleaving the carbon-sulfur bonds of recalcitrant thiophenic compounds contained in sour crude. We have simultaneously worked towards the completion of two proposed objectives: (objective 1) determining molecular-level contributions to substrate specificity and inhibition from active site dynamics and (objective 3) characterize the origins of substrate specificity through the reaction mechanism.
Molecular-level contributions to substrate specificity. We have completed molecular dynamics (MD) simulations of DszB bound with the substrate (2'-hydroxybiphenyl sulfinic acid, HBSP); the product (2-hydroxy biphenyl, 2-HBP); and several inhibitory compounds (Figure 1). The compounds represent three basic structural groups: functional groups on one side of the phenyl rings, on both phenyl rings, and planar naphthenic derivatives. This initial set of simulations was performed using unoptimized force field parameters, which does not provide the level of detail we require for this study. Thus, we began parameter optimization and validation to obtain the most accurate results possible from our simulations.
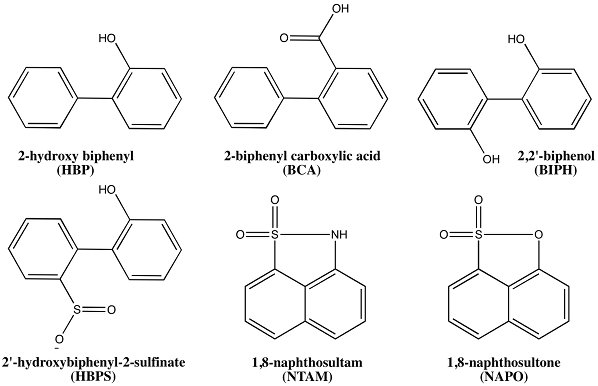
Figure 1. Aromatic analogs with varying inhibitory action on DszB. 2'-hydroxybiphenyl-2-sulfinate is the substrate, and 2-hydroxy biphenyl is the inhibitory product. 2-biphenyl carboxylic acid and 1,8-naphthosultone are non-inhibitory, yet not productive. 2,2'-biphenol and 1,8-naphthosultam inhibit DszB.
Using an established parameterization protocol compatible with the CHARMM potential energy function (U) (Equation 1), we have optimized and validated parameters for compounds in Figure 1. Potential energy parameters include: bond length (b); bond angle (θ); dihedral angle (ϕ); improper torsion angle (ψ); dihedral multiplicity and phase (n and dn, respectively); force constants for the bond, angle, dihedral angle, and improper dihedral angle (Kb, Kθ, Kϕ, and Kψ, respectively); Lennard-Jones well depth (ε) and distance at the minimum (Rmin,ij); distance between atoms (rij); partial atomic charge (qi or qj); and effective dielectric constant (εij). Equilibrium values are shown with subscripted zeros.
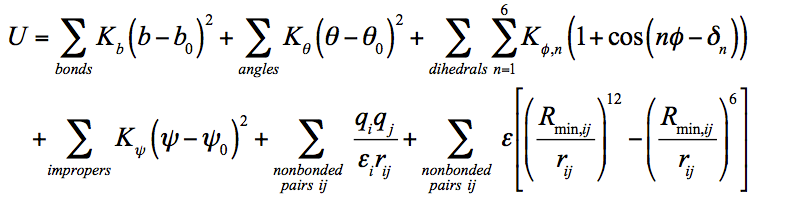 Eqn.
1
Eqn.
1
The parameters have been determined from optimized geometries obtained from quantum mechanical (QM) calculations using the MP2/6-31++G** basis set. Iterative parameter fitting was accomplished using the VMD Force Field Toolkit, with CGenFF parameters to initialize. Improper dihedral angle and Lennard-Jones nonbonded parameters were fixed. The fitting procedure – optimizing charges, angles, and then dihedral parameters – was repeated until the parameters converged.
The entire parameterization and validation procedure has been completed for each of the molecules above, and we are currently implementing these parameters in free energy calculations of DszB as described in the proposed research objectives. The accuracy of the parameters is illustrated in Figure 2, where the molecular mechanics (MM) and QM dihedral potential energy surfaces are compared; the contours and minima are well described.
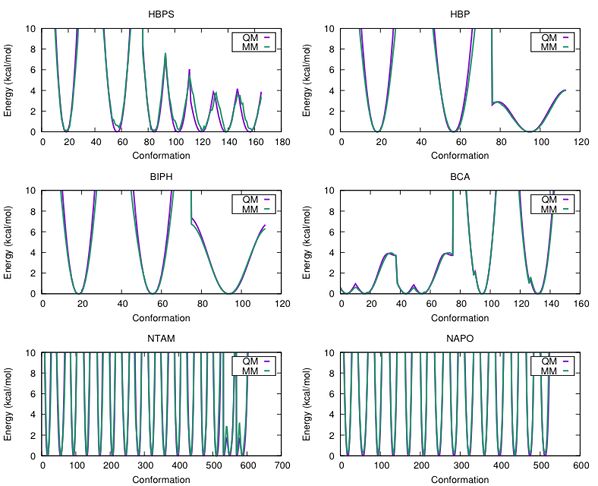
Figure 2. QM and MM dihedral potential energy surfaces for each of the parameterized molecules depicted in Figure 1. The x-axis represents the dihedral angle, ϕ, in degrees.
For all except HBPS, experimental infrared (IR) spectra are available for comparison to computed spectra (Figure 3). Using the new parameters, we calculated IR spectra for each compound in implicit solvent. In the low frequency range (<2000 cm-1), where aromatic rings or carboxylic acid functional groups are observed, the spectra are in agreement. Deviation of the computed spectra from experimental occurs in higher frequency regions (>2800 cm-1), where C—H and O—H stretching occurs. This is a known fault of calculations, where anharmonicity effects affect accuracy of calculated spectra. Overall, we have developed reliable force field parameters for small molecules of interest to the petroleum industry. These results will be submitted in manuscript form to the Journal of Molecular Graphics and Modelling and serve as the foundation for our future studies on substrate specificity of DszB.
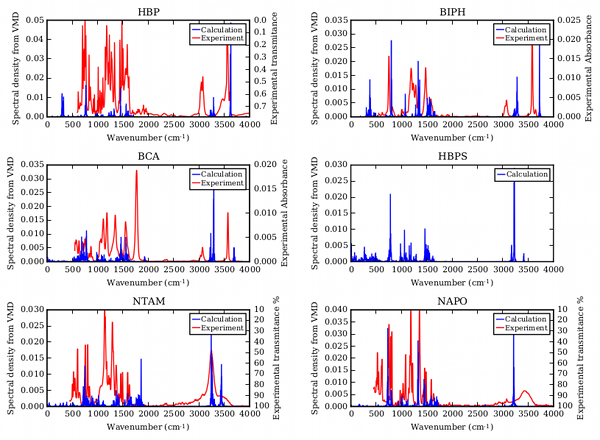
Figure 3. Comparison of experimental and calculated IR spectra validating force field parameters.
Characterization of the DszB reaction mechanism. The reaction mechanism was modeled using the cluster approach, in which the active site and important neighboring residues are cut from the enzyme and treated quantum mechanically. Steric and electronic effects of the protein environment are approximated by positional restraints and a homogeneous continuum medium, respectively. The crystal structure of the C27S variant of DszB in complex with HBPS (PDB ID: 2DE3) was used as the starting point for the calculations. Missing hydrogen atoms were added and the structure was minimized using MD (Figure 4a).
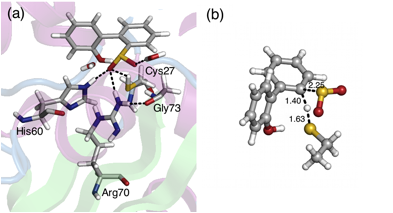
Figure 4. (a) Active site of DszB. (b) Transition state for desulfurization of HBPS by Cys27.
A model of the active site was built consisting of HBPS and the sidechain of Cys27 (truncated at Cα and saturated with H atoms), which is proposed to be the nucleophile in the reaction. Geometry optimization (with the position of Cα constrained) was performed in the gas phase at the B3LYP/6-31+G(d,p) level. Transition states were located by running potential energy scans along the distance between two reacting atoms. Two mechanisms were considered. The first involves the formation of a thiosulfonate-like intermediate in the first step through either (a) initial H atom transfer to one of the sulfinate O atoms or (b) concerted nucleophilic attack of Cys S atom on the sulfinate S atom and H atom transfer. For (a), no transition state was located and the resulting product, sulfinic acid, was found to be less stable than the reactant (8.3 kcal/mol higher in energy). On the other hand, the scan along the S-S reaction coordinate to locate a transition state for (b) resulted in dissociation of one of the sulfinate O atoms.
The second mechanism is the electrophilic substitution of the aryl sulfinate by the proton from Cys. A transition state was found (Figure 4b), which was confirmed by vibrational analysis to have one imaginary frequency. Intrinsic reaction coordinate calculations showed that the transition state leads directly to the release of SO2 from the substrate. The calculated free energy of activation for the reaction was 29.9 kcal/mol. Calculations are ongoing to investigate the effects of model size, functional, basis set, and solvation on the reaction barrier. We anticipate the results of this effort will be published as the first of two papers toward understanding the reaction mechanism of DszB.










