Reports: ND353962-ND3: Investigation of a Straightforward and Highly Customizable Synthetic Route to Triply Bidentate Phosphine Ligands
 printer friendly
printer friendlyThe first goal in our project was to develop a synthetic method to access heteroleptic tertiary phosphines, ideally using a method that avoids air- and water-sensitive intermediates. The proposed synthetic route was to convert phosphonates to phosphine oxides, followed by reduction of the phosphine oxides to the phosphines (Scheme 1). As described in this report, we were successful, and we reported the first facile and high yielding synthesis of phosphines directly from phosphonates.
Scheme 1. The proposed synthetic route from phosphonates to heteroleptic tertiary phosphines.
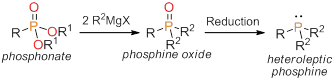
A variety of organometallic reagents were screened for the direct conversion of phosphonates to phosphine oxides. Grignard reagents proved to be the best candidates for this reaction while organo-zincates were non-reactive and organo-lithiates were too reactive, giving numerous undesirable products. Note that Grignard reagents are widely commercially available as well as easy to synthesize and handle, providing convenient methods for preparation.
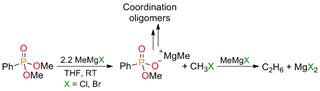
Our approach for the direct conversion of phosphonates to phosphine oxides also involved activating the phosphonate for nucleophilic attack. Lewis acid additives were screened in the expectation that they would increase the electrophilicity of the phosphonate through coordination – analogous to Lewis acid catalyzed nucleophilic ester substitution. After a rigorous Lewis acid screening, the highest yields were achieved with lutetium triflate (Lu(OTf)3). Although the yields were promising, they were still lower than desired. Experiments showed that the phosphonates were completely consumed in the reactions, demonstrating that a substantial side reaction was the major reason for the low yields. For example, in the reaction of MeMgBr with PhP(O)(OMe)2, a 23% yield of PhP(O)Me2 was obtained; however, 77% of the starting material in the reaction was unaccounted for by gas chromatography. These results suggested a non-volatile by-product is responsible for the low yields in the reaction. Further work showed that the side-reaction involved an Arbuzov-like decomposition of the phosphonates into alkyl halides and metal-phosphonate salts and oligomers (Scheme 2). We hypothesized that stoichiometric addition of a sodium salt should remove the Grignard halide from the reaction medium before it can decompose the phosphonate into the side-products. In fact, the addition of sodium triflate (NaOTf) to the reaction precipitated NaBr and essentially eliminated the side reaction. In consequence, the resulting yields of phosphine oxides were consistently high.
Scheme 2. Proposed steps and intermediates in the side reaction of PhP(O)(OMe)2 with Grignard reagents.
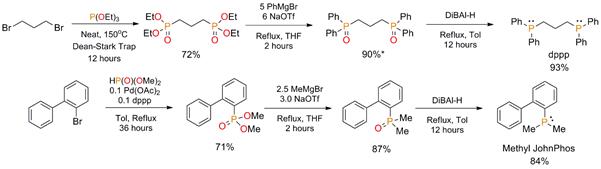
To highlight the usefulness of our new reaction method, the preparations of two phosphines were demonstrated. The classic and commercially available bidentate phosphine ligand 1,3-bis(diphenylphosphino)propane (dppp) was synthesized using nearly all benchtop techniques (Scheme 3a). The overall yield of the three-step synthesis was 60% starting from the alkyl bromide. Note that, although the commercial synthesis of dppp is well established, stoichiometric amounts of sodium metal are required. Also note that, although PhMgBr was used in this synthesis, other aryl Grignard reagents could also be used to make novel dppp derivatives that are not commercially available.
A new Buchwald-type ligand featuring a dimethyl phosphine moiety (Scheme 3b) was also synthesized. Dimethyl aryl phosphine derivatives are very laborious to synthesize, thus the traditional syntheses of similar phosphines rely on heavier and more electronically stabilized phosphorus precursors. For this reason, dimethyl-Buchwald-type ligands have, to the best of our knowledge, never been synthesized. As shown in Scheme 3b, the first synthesis of [1,1'-biphenyl]-2-yldimethylphosphine or “Methyl JohnPhos” was achieved in a three-step reaction with a 52% overall yield using inexpensive chemicals starting from the aryl bromide. The reaction is also easily scalable.
Scheme 3. a) Synthesis of dppp using the phosphonate to phosphine oxide single step reaction as the key step (*isolated as the mono-hydrate.) b) Synthesis of Methyl JohnPhos, a new Buchwald-type ligand. All yields are for isolated product.
In summary, a new method to synthesize phosphines directly from phosphonates was developed. The method utilizes all air-stable intermediates, readily available starting materials, Grignard reagents for direct P-C functionalization, and it produces high yields, all of which are advantageous compared to traditional synthetic routes. Note that, although the choice to use aluminum-hydride reagents (Scheme 3) as reducing agents was one made out of synthetic convenience, there is a considerable body of literature reporting alternative reducing agents that are mild and selective for converting phosphine oxides to phosphines. Overall, the method provides an extremely mild route for the synthesis of targeted phosphines. Finally, note that a diverse array of functionalization can be appended using any number of commercially available or custom-made Grignard reagents.










