Reports: ND653264-ND6: Thermodynamic Insights into Surfactant Assembly in Protic Ionic Liquids
Henry Ashbaugh, PhD, Tulane University
In year 2 we have considered
the thermodynamics of dissolution of methane in the ionic liquid/water mixtures
described above as well as begun to look at the structure of ionic liquid
mixtures with ethyl alcohol. Our studies of methane
dissolution in the ionic liquid mixtures is intended to provide insights
into the thermodynamics of nonpolar solute solvation which is ultimately
believed to drive assembly surfactant assembly. Our studies of the structure of
mixtures of ethanol with the ionic liquids is intended to examine aggregation
of short amphiphilic species (ethanol) in these
solvents, which have been observed from scattering experiments to exhibit a
broad range of large scale structures.
Our initial studies of
methane solvation focused on mixtures of 0 vol%, 25 vol%, 50 vol%, 75 vol%, and 100 vol% ionic liquid
with water. Each mixture was simulated over a broad temperature range using
replica exchange molecular dynamics at 1 atm. Methane excess chemical
potentials were determined using standard particle insertion techniques.
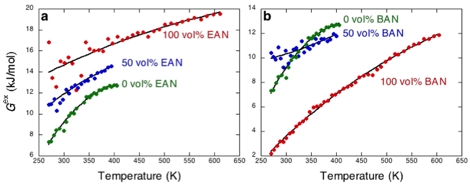
Figure
1.Excess
chemical potential (Gex)
of methane in mixtures of EAN with water (a)
and BAN with water (b) as a function
of temperature at atmospheric pressure. Points are simulation results,
while the solid lines indicate fits to the Margules
expansion.
The excess chemical
potentials of methane in aqueous EAN and BAN mixtures are shown in Figure 1. Both
plots indicate that the chemical potential is positive in these mixtures,
indicative of low gas solubility. Moreover methane's excess chemical potential
is an increasing function of temperature, indicating that dissolution is entropically unfavorable in both solvent mixtures.
Interestingly the excess chemical potential appears to approach a maximum in
pure water (green points), while is does not do so in the other solvents. This
maximum indicates both a change in the sign of the solvation entropy in pure
water that results from a significant large positive heat capacity for dissolution
in water. Therefore, while methane solvation in these solvents is entropically unfavorable (a characteristic of nonpolar
solvation in water, but not normal organic solvent) in all solvents, solvation
in water still exhibits distinct temperature dependences not observed in the
ionic liquids.
The excess chemical potential
of methane in pure EAN is great than that in pure water (Figure 1a), indicting
a lower solubility for methane in the ionic liquid. This result is surprising
since experiments generally find the opposite trend. One potential reason for
this discrepancy is the lack of polarizability in the
model. We have demonstrated that the effective inclusion of polarization for alkane
solvation in water through inclusion of an increased water/solute attractive
interaction can account for discrepancies between experiment and simulation for
simulation model parameterized to pure hydrocarbon properties. In the case of
nonpolar solvation in an ionic liquid we may expect an even greater
contribution from polarization due to the large electrostatic fields in the
charged solvent. As the alkyl length of the ionic liquid grows, methane's solubility
increases as indicated by the lowering of the chemical potential. Indeed for
BAN methane's solubility is greater than in water (Figure 1b). We therefore
expect that the driving force for assembly is greater in EAN than BAN. It is
generally reported that assembly is weakest in BAN in agreement with the result
reported in Figure 1.
To correlate the excess
chemical potential of methane in these mixtures we employed a Margules-like expression for the free energy
wherex1
is the mole fraction of component 1 (taken to be the ionic liquid here), T is
the temperature, and A and B are Margules
constants. The temperature dependence of the terms Gex(T, x1=1), Gex(T, x1=0),
A(T),
and B(T) were assumed to follow the standard form X(T) = a + b*(T-300K) + c*Tln(T/300K), where a, b, and c are fitted coefficients. Fits of this expression are indicated by the solid lines in Figure
1. Overall this expression does an excellent job allows us to
interpolate to solvent compositions that were not simulated.
In x-ray scattering study of
alcohols in protic ionic liquids, Warr's
group (J PhysChem, 2014)
observed large scale structuring of ethanol and longer chain alcohols in EAN
and PAN mixtures. In their study they observed an increase in scattering over a
wide range of length scales that was not present for either the EAN, PAN, or
ethanol alone.
These results suggested the
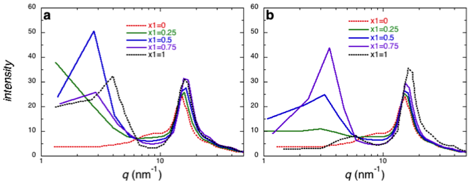
formation of self-assembled aggregates with a broad range of sizes. In figure 2
we show predicted x-ray scattering structures obtained from our simulations of
ethanol mixtures with EAN and BAN.
Figure 2. Predicted x-ray scattering intensity from
molecular simulation for mixtures of EAN with enthanol
(a) and BAN with ethanol (b). The mole fractions (x1) reported in
the figure legends correspond to the mole fraction of the ionic liquid in the
mixture.
These simulations clearly
show the onset of larger scale structures in both ionic liquids as indicated by
the increase in scattering at low q
in these figures. While EAN itself exhibits large scale
structure, this peak shifts to larger scale structures in mixtures with
ethanol. Given the low solubility of methane in EAN it is possible that this
structure results from phase separation. When we examine molecular snapshots
from these simulations, the structure appears more like a bicontinuous
sponge. We have raised the temperature significantly to try to melt out these
structures and permit phase separation if that is the equilibrium state. The
observed simulation and x-ray structures, however, we resilient to heating
suggesting we are sampling the true equilibrium state for our simulations. That
said, the experimental data from Warr indicated a
broad range of aggregate sizes even greater than what we observe. We are
limited in our simulations to structures smaller than ~5 nm. While the ethanol
to induce structuring in BAN (Figure 2b), the scattering intensity is not as
pronounced as in EAN. This is consistent with observations above than methane
is more soluble in BAN, thereby reducing the driving force for assembly.
 printer friendly
printer friendly