Reports: ND652100-ND6: Multiscale Investigation of Asphaltene Self-Assembly
William G. Noid, PhD, Pennsylvania State University
Crude
oil is an extremely complex mixture of petrochemicals. Asphaltenes form the heaviest and most
aromatic component of this polydiverse mixture. In
practice, asphaltenes are operationally defined as a solubility class that is
soluble in toluene and insoluble in n-heptane. It is believed that asphaltene molecules
form nanoaggregates at very low concentrations and
that these nanoaggregates exist as a stable emulsion
in crude oil. However, when this
emulsion is destabilized, asphaltenes form macroscopic aggregates that create
tremendous problems for the petrochemical industry. For this reason, asphaltenes have been
nicknamed the cholesterol of petroleum.
Molecular
dynamics simulations provide a powerful computational tool for investigating
the interactions, structure, and thermodynamic stability of asphaltenes and
their aggregates. Unfortunately,
conventional atomically detailed models remain prohibitively inefficient for
investigating the relevant length and time scales that are industrially
relevant for asphaltene aggregation.
Because they are far more efficient, lower resolution coarse-grained
(CG) models provide a far more promising means for simulation studies of
asphaltene aggregation. However, it
remains challenging to develop CG models that accurately model both structure
and thermodynamic properties.
In particular, bottom-up models typically provide a more accurate
description of structural properties, while top-down models generally provide a
more accurate description of thermodynamic properties.
During
the past funding period, we have continued our efforts to develop bottom-up CG
models that accurately model both structure and thermodynamic properties. By extending the previous work of Das
and Andersen (DA), we (Dunn and Noid, DN) have demonstrated that rigorous,
bottom-up approaches can accurately describe the structure, average density,
and compressibility of atomically detailed models for industrially relevant
asphaltene solvents. The first
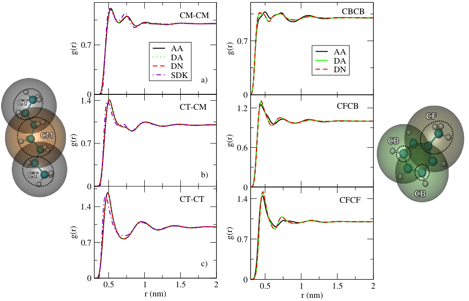
figure below compares for three site models of heptane (left) and toluene
(right), the site-site radial distribution functions (rdfs)
sampled by an atomic model (AA), with the rdfs
sampled by the DA and DN bottom-up models.
In each case, the bottom-up model quite accurately reproduces the AA
structure. In contrast, a
comparable 3-site top-down model (SDK) for heptane provides a less accurate
description of the AA structure.
Figure 1: Comparison of site-site
radial distribution functions (rdfs) for heptane
(left) and toluene (right). In each
panel, the solid black curves represent rdfs for the
OPLS-AA model, while the green and red curves present rdfs
for the DA and DN bottom-up models.
In the right column, the dashed-dotted purple curve presents rdfs for the top-down SDK model.
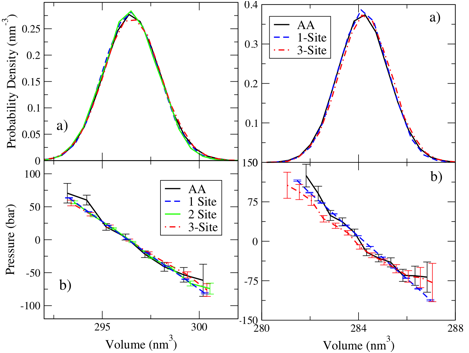
More
importantly, the following figure demonstrates that the resulting bottom-up
models accurately reproduce the density fluctuations, which reflect the
equilibrium liquid density and compressibility, as well as the pressure
equation of state (near the simulated conditions of 1-bar pressure) for the
atomic models. In particular, this
figure demonstrates that the CG models reproduce these thermodynamic properties
with similar accuracy for 1, 2, and 3-site heptane models (left) and 1 and
3-site toluene models (right). This
is particularly important since it paves the way for performing simulation
studies of asphaltenes at a variety of different resolutions.
Figure 2: Density fluctuations and simulated
pressure equations of state for heptane (left) and toluene (right). In each panel, the solid black curves
represent results for the OPLS-AA model, while the blue, green, and red curves
present rdfs for 1, 2, and 3-site DN bottom-up
models.
We
are currently finalizing results that appear to demonstrate similar success for
a transferable bottom-up model of heptane-toluene mixtures at a range of
compositions that vary from pure heptane to pure toluene. Our initial work focused on these
particular systems because asphaltenes are operationally defined on the basis
of their relative solubility in toluene and heptane. By applying the methodology that we have
developed during past funding periods, we are also working to parameterize
bottom-up CG models for model asphaltene compounds in these solvents.
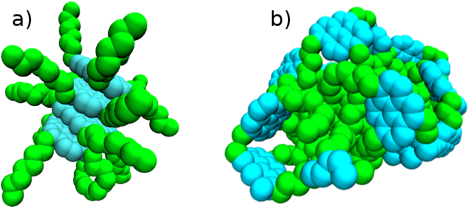
In
addition, we have also initiated simulation studies with phenomenological
top-down models of model asphaltene compounds. As indicated below, preliminary results
of these studies demonstrate the impact of solvent quality and asphaltene
architecture upon the nanoaggregates that form. The insights from these studies with
top-down models will guide future investigations with bottom-up models that
more accurately model asphaltene-solvent interactions.
Figure 3: Representative asphaltene nanoaggregates that form under different simulated
conditions.
 printer friendly
printer friendly