Reports: DNI655801-DNI6: Understanding the Effect of Molecular Topology on Surfactants for Enhanced Oil Recovery
 printer friendly
printer friendlyThe goal of this project is to gain a fundamental understanding on the effect of surfactant topology and chemistry on the properties that directly influence its suitability for CO2 flooding applications, including the surfactant's ability to reduce the interfacial tension (IFT) of CO2/water interfaces. During the period of this report, we developed software to automatically explore the space of surfactant tail topologies. Simulations were carried out using a coarse-grained (CG) model based on the SAFT-γ equation of state [Lobanova et al., J. Phys. Chem. B 115, 11154 (2011)], and were used to estimate the surface tension of water-air systems at the critical micellar concentration (CMC) for over 60 surfactant designs. Prior experimental studies have found that the molecular packing of tail groups and surface tension of air-water interfaces correlate strongly with the ability of the surfactant to lower the surface tension of CO2/water interfaces [Mohamed et al., Langmuir 28, 6299 (2012)], and studying this system enabled us to scan a large topology space rapidly.
The surfactant model used for the topology scans consists on two hydrophilic head group beads parameterized to reproduce the alcohol terminus of an ethoxylated surfactant, and tail group beads parameterized to reproduce properties of the alkane series. Water was modeled using beads comprising two water molecules per bead (Figure 1). We validated the model by comparing with experimental values for the IFT of water/air, decanol/air, and water/air interfaces containing linear ethoxylated surfactants. In parallel with these calculations, we have been developing an extended ionic surfactant model based on the Shinoda, DeVane, and Klein (SDK) force field, as explained below, and are currently investigating the effect of surfactant topology for IFT reduction in brine/air interfaces.
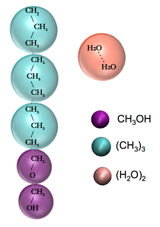
Figure 1. An illustration of the CG model surfactant used to predict the effectiveness of different surfactant designs.
Some of results, showing the effect of surfactant topology on the IFT of water/air interfaces at the CMC for surfactants with different tail group sizes, are summarized in Figures 2, 3, and 4. The results show a complex relation between surfactant effectiveness and tail group topology. The main general observations are:
- Linear surfactants are consistently among the worst in comparison to their branched counterparts.
- Surfactants with longer branches generally outperform those with short branches.
- Having a higher branching density close to the head group generally improves surfactant effectiveness.
We are currently extending our data to cover a group of over 100 surfactants, and will correlate the data using a set of molecular topology descriptors. This will be useful as a tool to rapidly test surfactant designs and pinpoint those with high potential effectiveness.
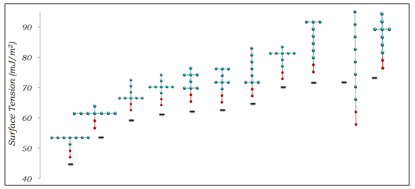
Figure 2. IFT of water/air interfaces at the CMC for surfactants containing 8 CG tail group units.
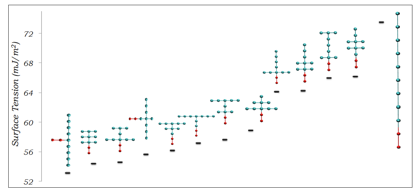
Figure 3. IFT of water/air interfaces at the CMC for surfactants containing 9 CG tail group units.
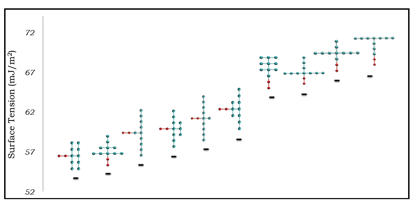
Figure 4. IFT of water/air interfaces at the CMC for surfactants containing 10 CG tail group units.
In addition to modeling ethoxylated surfactants, this year we have worked on an extension of the SDK [Shinoda et al., Soft Matt. 4, 2454 (2008)] model to allow for testing different ionic surfactant architectures in the same manner. In order to obtain mixing parameters for the additional functional groups not included in their work, we will extend an approach proposed by Haslam et al., where the pair interactions are estimated from the ionization energy, dipole moments, and quadrupole moments of the molecular fragments [Haslam et al., Fluid Phase Equil. 266, 105 (2008)]. Since their model was derived for the SAFT-VR equation, we will reproduce their steps using a Mie potential in order to derive the mixing rules. Since the ionization energy and dipole moment of molecular fragments are not experimentally accessible values, we will estimate them using electronic structure calculations. As for the level of theory, we have tested the B3LYP, MP2, and wB97XD models with multiple basis sets to compare their accuracy to experimental data. B3LYP and MP2 were chosen because of their common use and wB97XD for its use in calculations requiring diffuse basis sets [Orestes et al., Phys. Chem. Chem. Phys. 16, 17213 (2014)], as is our case with the ionic beads. The basis sets compared were combinations of polarized and diffuse functions added to the 6-31G, Dunning's correlation consistent, and Ahlrichs and coworkers def2 basis sets. The results showed that the MP2/def2TZVPD combination provided the necessary level of accuracy while providing sufficient computational efficiency. These methods will be used to estimate the mixing parameters using the procedure described above.










