Reports: DNI154824-DNI1: Site- and Enantioselective Electrophilic Halogen-Transfer Processes
 printer friendly
printer friendlyTwo powerful approaches promise to transform strategic small molecule assembly: late-stage atom-transfer processes and cross-coupling reactions. Both of these types of transformations enable previously unfeasible retrosynthetic disconnections, and this grant has supported advances related to both of these technologies. We have specifically targeted the development of new pre-catalysts to enable previously infeasible fluorination reactions. To this end, assays have been developed to study fluorine installation into sp3-hybridized carbon centers and the competitive oxygen-atom transfer process. With these assays in hand, we have interrogated previously reported fluorination methods to investigate the efficiency of fluorine transfer with a slightly broader range of metal-ligand complexes. Additionally, in the course of these efforts, we have developed selective, serial and exhaustive Suzuki-Miyaura reactions of polychloropyridines with alkylpinacol boronic esters. Consequently, this grant has laid the foundation to enable currently unfeasible fluorination processes, and has streamlined important and previously challenging cross-coupling reactions.
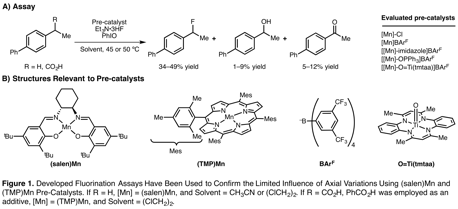
Assay for Fluorination and Oxygenation. Investigations began with the development of proton NMR assays to document the efficiency and selectivity of halogenation of benzylic substrates (Figure 1). These assays have been validated by reproducing previously disclosed reactions. To evaluate the impact of axial donor ligands on fluorination, we employed novel pre-catalysts that differed from each other based on the incorporation of a neutral axial donor ligand. When these novel pre-catalysts were compared with known catalysts, they achieved similar results in the fluorination reaction. These observations are consistent with previous mechanistic proposals in which fluoride serves as the axial ligand in the active catalyst system. Ongoing efforts focus on identification of alternative modular catalyst architectures to enable halogen installation in place of an sp3-hybridized carbon-hydogen bond with improved efficiency and selectivity.
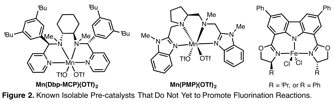
Evaluation of Modular Catalyst Architectures. To extend this line of research, we have identified, synthesized and evaluated a series of catalyst architectures that have not been disclosed for use in fluorination reactions. Thus far, these catalyst architectures have favored oxygenation relative to halogenation (Figure 2). Consequently, the accessed pre-catalyst architectures are being evaluated in other atom-transfer reactions.
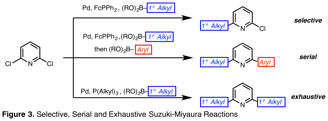
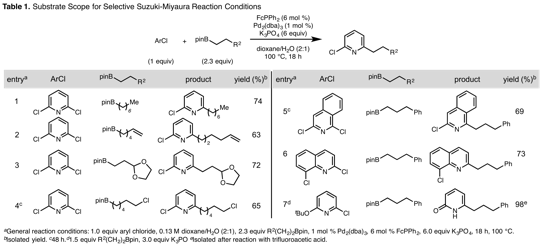
Selective, Serial and Exhaustive Suzuki-Miyaura Reactions of 2,6-Dihalopyridines with Alkylpinacol Boronic Esters. Initially, this series of methods was investigated during the course of efforts to access a catalyst scaffold for use in atom-transfer reactions. The envisioned route access a pyridine-centered ligand based on mild cross-coupling strategies. We identified the Suzuki-Miyaura reaction for the proposed route due to a series of practical advantages, including mild reaction conditions, commercially available reagents with low toxicity, and broad functional group tolerance. Nevertheless, we found few conditions that could be used to cross-couple alkyl organoboron compounds with aryl halides, especially 2-pyridyl halides. We have developed and disclosed two novel Suzuki-Miyaura protocols (Org. Lett. 2016, 18, 4440-4443). Under the disclosed conditions, polyhalogenated six-membered heteroarenes undergo cross-coupling reactions with predictable selectivity, or react with distinct organoboron compounds to form two new carbon–carbon bonds in series in a single reaction vessel (Figure 3). As an undisclosed extension, a third set of conditions have been identified to permit exhaustive cross-coupling of 2,6-dihalopyridines with alkylpinacol boronic acids.
Under the optimal conditions for the selective transformation, 2,6-dichloropyridines react with alkyl boronic pinacol esters with K3PO4 as a base, 1 mol % Pd2(dba)3, and 6 mol % FcPPh2 in 2:1 dioxane:H2O at 100 °C to afford desired 2-chloro-6-alkylpyridines in excellent selectivity and good isolated yield (Table 1, entry 1). This transformation tolerates alkyl pinacol boronic esters that contain distal functional groups including an olefin, an acetal, and an alkyl chloride (entries 2-4). These conditions also enable the selective cross-coupling of 1,3-dichloroisoquinoline at the more sterically encumbered C(1) position (entry 5). This selectivity complements previously disclosed conditions, which preferentially install an aryl group at C(1).
We have merged this selective method with a compatible aryl-aryl cross-coupling reaction to demonstrate the practical utility of preserving a pyridyl C–Cl bond. The resultant transformation relies on the faster transmetallation of arylboron compounds relative to alkylboron reagents. The developed transformation cross-couples a variety of arylboron compounds, including arylboronic esters and acids, N-methyl-iminodiacetic acid boronate esters, and trifluoroborate salts (Table 2, entries 1–4). The one-pot method transforms phenyl, pyridyl, thiophenyl and furyl boron compounds (entries 1–7) to generate a series of 2-alkyl-6-arylpyridines in good isolated yields.
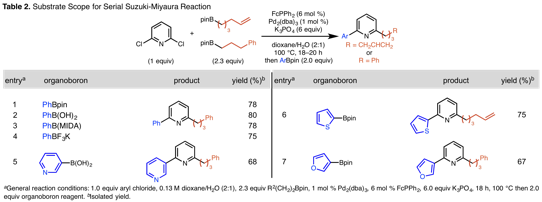
These disclosed methods merge air stable pinacol boronic esters with cheap, readily available chloroaryl compounds. The chemoselective transformation predictably transforms polychlorinated substrates to monoalkylated pyridyl or benzyl products. Additionally, a one-pot cross-coupling sequence incorporates alkyl- and then arylboron reagents to access differentially substituted pyridyl motifs. These conditions provide efficient access to high value small molecules.
Impact of Funding on Co-workers. Each of the talented individuals funded by this ACS PRF grant gained research experience with ligand synthesis, organometallic catalyst development, and methodology development. Importantly, this research grant provided stipend support Dr. S. Laulhe in his ultimate year as a postdoctoral scientist, and the research he carried out enabled him to secure a tenure-track faculty position in the Department of Chemistry and Chemical Biology at Indiana University-Purdue University Indiana. In addition, this grant partially supported the work of graduate student Mina Shehata (partial stipend), undergraduate students Ekaterina Khlystova (partial summer stipend) and Ashling Zhang (supplies). For Ms. Zhang, this support enabled her first synthetic research experiences as an independent study student.
In the course of this research, trainees acquired experience as scientific communicators. All trainees have presented their work in group meetings. At Duke, Ms. Zhang presented her ligand synthesis at the Spring 2016 Visible Thinking poster session, and Mr. Shehata is in the process of preparing a poster for the Chemistry Department Graduate Research Symposium. In more formal disclosures, Dr. Laulhe wrote the first draft of the manuscript to disclose selective and serial Suzuki-Miyaura reactions of 2,6-dichloropyridines, and presented his research at the Organic Reactions and Processes Gordon Research Conference.










