Reports: ND554684-ND5: Spectro-Electrochemical Microscopy of Single Nanoparticle Electrocatalysts
 printer friendly
printer friendlyThe objective of this project is to correlate the catalytic activity of gold nanoparticle electrocatalysts with nanoparticle morphology and surface chemistry for the reduction of CO2 using single particle spectro-electrochemical microscopy. Our project’s central hypothesis is that the catalytic activity of gold nanoparticle electrocatalysts strongly depends on size, shape, crystal facet and surface chemistry, and that possibly only a few catalytic 'hot spots' dominate the signal in ensemble measurements. The project goals are: (1) Establish the relationship between the electron density of a single gold nanoparticle and the induced spectral changes of the plasmon scattering response (wavelength maximum, intensity, linewidth) as a function of nanoparticle morphology. (2) Determine the catalyst activity for the CO2 reduction using different gold nanoparticle electrocatalysts. (3) Identify the spatial extent of active catalytic sites on single gold nanoparticles using super-resolution electrochemical luminescence (ECL) microscopy.
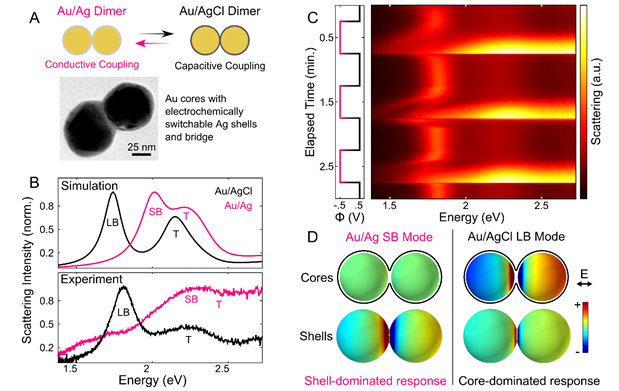
Our second year of funding has led us in new exciting directions because of discoveries made along the way. The primary motivation for our new project is that, because the optical properties of metallic nanoparticles, particularly when in close proximity to each other, are highly sensitive to surface chemistry, it should be possible to use the plasmon resonance frequency, linewidth, and intensity to monitor catalytic processes occurring on the nanoparticle surface. Our latest results demonstrate that, by monitoring the plasmon response, we can achieve reversible oxidation - reduction chemistry of Ag - AgCl shells on the surfaces of both individual and strongly coupled Au nanoparticle pairs, resulting in extreme but reversible changes in scattering line shape. By reversibly tuning the redox chemistry of Ag shells on strongly coupled Au nanoparticle dimers, we are the first group to demonstrate reversible formation of the charge transfer plasmon mode via switching between capacitive and conductive electronic coupling mechanisms. (Fig. 1)
Dynamic single particle spectroelectrochemistry also gives insight into the reaction kinetics and evolution of the charge transfer plasmon in an electrochemically tunable structure. Our study represents a highly useful approach for precise tuning of the morphology of narrow interparticle gaps, and will be of value for controlling and activating a range of properties such as extreme plasmon modulation, nanoscopic plasmon switching, and sub-nanometer tunable gap applications.
Fig. 1. Reversible electrochemical tuning of a dimer between capacitive and conductive coupling. (A) Overlapping shells on Au dimers were switched between Ag and AgCl to create a switch between capacitive and conductive coupling. TEM image shows Au dimer enveloped by Ag shell. (B) Simulated and experimental scattering spectra show drastic optical response to switching plasmon coupling mechanism. (C) Series of scattering spectra during step potential application to switch shell composition. (D) Surface charge density plots of shell and core surfaces for screened bonding (SB) and longitudinal bonding (LB) modes. Charge plots were calculated at 2.0 eV for the SB mode and 1.67 eV for the LB mode. |
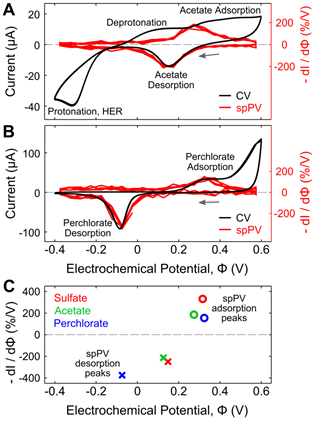
One of the most important physical processes that must occur for heterogeneous photocatalysis is surface adsorption. This year we also established means of reversibly inducing and monitoring the adsorption of polyanions to gold nanoparticle surfaces. We have developed single-particle plasmon voltammetry (spPV) using plasmonic nanoparticles. spPV can be used to detect sulfate electroadsorption to individual Au nanoparticles. By comparing both semiconducting and metallic thin film substrates with Au nanoparticle monomers and dimers, we demonstrated that using Au film substrates improved the signal in detecting sulfate electroadsorption and desorption through adsorbate modulated thin film conductance. Using single-particle surface plasmon spectroscopic techniques, we constructed spPV to distinguish between sulfate, acetate, and perchlorate adsorption on coupled Au nanoparticles. (Fig. 2)
|
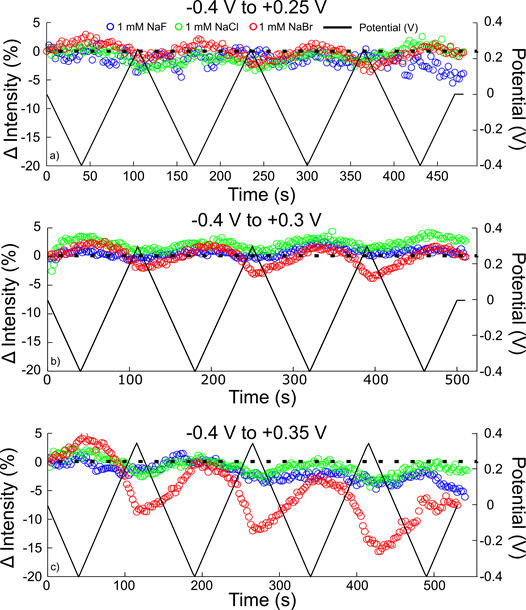
Finally, heterogeneous photocatalysis on highly reactivity of plasmonic nanoparticles can result in a range of side processes including catalyst poisoning, surface etching, or dissolution. Such processes have the potential to either improve or worsen catalytic turnover. Thus, our final efforts in the past year were aimed at understanding such processes. A spectroelectrochemical flow cell was used to probe the localized surface plasmon resonance of the same single gold nanorods in sodium fluoride, sodium chloride, and sodium bromide electrolytes using dark-field scattering. The changes in resonance energy, linewidth (full-width at half-maximum, FWHM), and peak intensity of a Lorentzian fit to single nanorod scattering spectra as the rods charge were compared to determine the role of anion adsorption in changing the nanorod surface chemistry. We demonstrated that at positive potentials up to +0.25 V, the induced changes in the LSPR were independent of halide anion. At more positive potentials (+0.3 V to +0.35 V) bromide and chloride ions damped the signal, observed as an increase in the linewidth. At the most positive potential investigated in all three electrolyte solutions (+0.35 V), the AuNR scattering intensity decreased in bromide electrolyte, indicating dissolution (Fig. 3). The kinetics of the bromide-mediated dissolution were shown to be controlled by the electrolyte concentration and show that the rate of dissolution increases with each cycle from negative to positive potential.
Fig. 3. Reversibility of change in intensity as a function of potential range. Change in intensity relative to the initial 0 V measurement for three different single AuNRs from -0.40 V to +0.25 V (a), -0.40 V to +0.30 V (b), and -0.40 V to +0.35 V (c) in 1 mM NaF (blue), NaCl (green), and NaBr (red) electrolyte. The applied potential cycle (black) is superimposed over the scattering intensity. The dotted black line represents no change from the initial intensity.
|