Reports: DNI353764-DNI3: Copper Photosensitizers for Photoredox Catalysis
 printer friendly
printer friendlyIn continued efforts to understand light-driven chemistry, work in our lab during the second year of this project has focused on examining how proton transfer reactions (acid-base chemistry) can be activated with light. We recognize that proton transfers are important components of many organic transformations and proton-coupled electron transfer processes in general, but little is understood about these reactions especially in non-aqueous (organic) solvents. Moreover, it is not clear how to these reactions can be promoted with visible light, which is key for applications like photoredox catalysis and light-driven fuel production. Specifically we have aimed (1) to develop systems with optical handles of proton transfer so that we can track these reactions spectroscopically and (2) to understand excited-state acid-base chemistry better, with a focus on photobasicity.
Over the last year, we have examined the thermal and excited-state reactivity of 7-(dimethylamino)quinoline derivatives (R-7DMAQ). These molecules have optical signatures in the visible region and shift upon protonation (to form R-7DMAQH+) (Fig. 1). This bathochromic shift provides an optical handle to optically monitor proton transfer as a function of time using methods like stopped-flow rapid mixing and transient absorption spectroscopy. Furthermore, substitution at the 2- and 4- positions of the quinoline ring allows the pKa (in CH3CN) to be systematically tuned and thus proton transfer driving force to be modulated.
Thermal reactions were first explored in order to examine how these quinolines could be employed as “photometric bases” to improve our understanding of proton transfer in non-aqueous solvents. The proton-coupled electron transfer oxidation of p-aminophenol was chosen as a model reaction; oxidation of p-aminophenol by the mild oxidant ferrocenium in the presence of the 7-DMAQ yielded the initial products of ferrocene, the quinolinium (7-DMAQH+) and the phenol radical cation (NH2PhOH●+). The electron transfer component was optically monitored by tracking the loss of the ferrocenium signal at 620 nm, and proton transfer was tracked by monitoring the rise of the quinolinium signal at ca. 450 nm. The initial reaction pathway was diagnosed as a stepwise ET-PT reaction, followed by subsequent PCET reactivity of the NH2PhOH●+ product. Kinetics modeling was employed to determine rate constants for the elementary steps, including proton transfer. These data revealed an intriguing free energy relationship for proton transfer—clear correlations were observed between kPT and the pKa of the conjugate acid of the quinolinium, which is directly proportional to driving force for proton transfer. The data are best interpreted by a Marcus relationship for proton transfer, adding to the very small family of experimental examples in which this long-theorized relationship has been observed (Fig. 2).
The excited-state reactivity of these 7-(dimethylamino)quinoline derivatives has revealed that these complexes are photobases—molecules that become more basic upon electron excitation (pKa* > pKa, Fig. 3). While the photophysics behind photoacidity are generally understood, less is known about photobasicity. We discovered that the excited-state acid-base reactivity of these quinolines is influenced by both electronic and structural factors. Excitation with visible light in the presence of acids (in CH3CN) revealed that the fluorescence is rapidly quenched. Transient absorption spectroscopy was utilized to confirm that quenching proceeded via excited-state proton transfer to the quinoline, as the spectroscopic signature for the quinolinium product could be readily resolved. The subsequent thermal back proton transfer was also examined through transient absorption spectroscopy. The data together reveal that excited-state proton transfer dynamics are influenced by more than just driving force for proton transfer—electronic and structural factors play an important role. By contrast, thermal proton transfer reactions appear to exhibit clear free energy relationships. These results are guiding our understanding of how light can be used to promote proton transfer reactions.
Together these studies have provided new understanding of the factors influencing proton transfer in organic solvents and revealed opportunity to exploit the optical signatures of photometric bases to examine the reaction dynamics of thermal and excited-state proton transfer reactions.
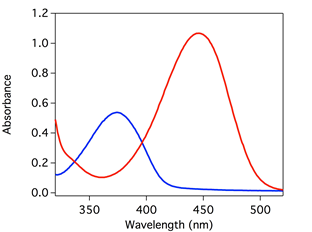
Figure 1. UV-vis absorbance spectrum of a 7-(dimethylamino)quinoline and its conjugate acid in acetonitrile.
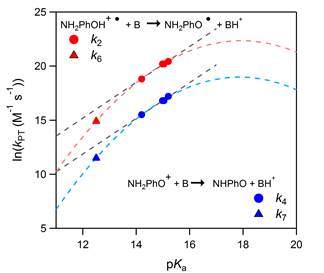
Figure 2. For proton transfer reactions from aminophenol oxidation products, free energy relationships are observed between the rate constants and the pKa value for the conjugate acid of the proton acceptor, which is proportional to the driving force for proton transfer within each set of reactions.
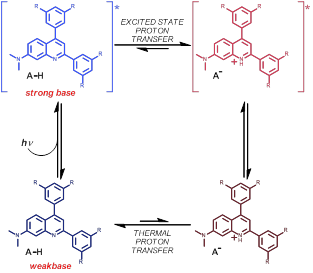
Figure 3. Excited-state proton transfer of quinolines is well-described by the Forster cycle.










