Reports: UNI354581-UNI3: Investigating Novel Transition Metal Complexes for Use in Asymmetric Catalysis
 printer friendly
printer friendlyBackground
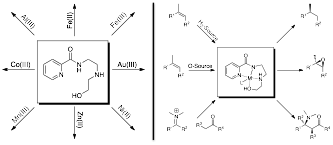
Figure 1. Proposed synthesis of chiral-at-metal coordination complexes with a tetradentate ligand (left); and examination of those chiral-at-metal complexes in catalysis (right).
Inspired by the pioneering Nobel Prize winning work of Knowles, Nayori, and Sharpless in asymmetric catalysis, and encouraged by the subsequent advances of Jacobsen and others with metallosalen catalysts, we proposed to synthesize a variety of chiral-at-metal metal complexes and test their activity as Lewis acid catalysts. The proposed synthesis is facile, and crystallography of the first of these compounds, a Cu-complex, demonstrated an approximately square pyramidal geometry, like that of the metallosalen complexes. Given the free coordination site in this geometry, it is hypothesized that such complexes should make excellent Lewis acid catalysts. Moreover, if catalytic, these compounds would be predicted have tremendous potential for asymmetric catalysis given the ability to tune their reactivity by modifying the ligand around the central metal.
Results:
In the first report we described our success synthesizing, structurally characterizing, and testing as Lewis acid catalysts several chiral-at-metal complexes with a tetradentate ligand. In this report we describe our progress in several directions. First, we attempted to generate more Lewis acidic catalysts by dehalogenation of our first generation metal complexes of cobalt (2) and copper (3) using silver (I) nitrate (Figure 2).
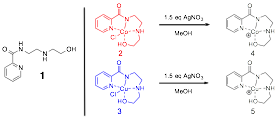
Figure 2. Proposed dehalogenation of complexes 2 and 3.
We hypothesized that these formally positively charged complexes would be more reactive by virtue of their greater Lewis acidity. We then structurally characterized the metal complexes produced by these dehalogenation reactions as well exploring metal complexes from Mn(II), Mn(III), Ni(II), Fe(III), Cu(I), and Zn(II). Of these metals, the only discrete complex that were isolated in good yield were those made with Ni(II) and Zn(II). The Zn(II) complex was isolated crystallization from the neat reaction mixture and characterized by X-ray crystallography (Figure 3).
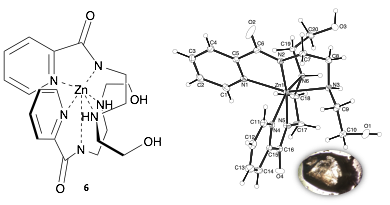
Figure 3. Zn complex (6) formed by reaction of ligand 1 and ZnCl2 (left); ORTEP representation of compound 6 crystals with 50% probability thermal ellipsoids (right).
Ultimately, dehalogenation of complexes 2 and 3 did not produce the desired products, instead forming additional coordinative interactions with alternative ligands (Figure 4). It seems most likely that the additional ligand 1 necessary to form complex 8 results from a decomposition of complex 3 following dehalogenation. This semi-labile species may prove an effective catalyst, although we have not yet adequately exhausted this possibility. Complex 8 was examined as a chiral catalyst, although complex 7 was not because it lacks available coordination sites and is therefore unlikely to act as an effective Lewis acid catalyst.
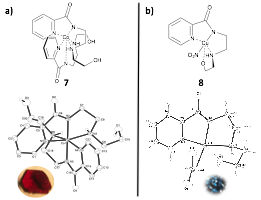
Figure 4. (a) Two-dimensional and ORTEP representation of compound 7 crystals with 50% probability thermal ellipsoids, and; (b) Two-dimensional and ORTEP representation of compound 8 crystals with 50% probability thermal ellipsoids formed by the silver nitrate promoted dechlorination of complexes 2 and 3 respectively. In both cases, chloride cannot be removed without being replaced by an additional ligand.
We hypothesized that these undesired coordination reactions could be avoided by stabilizing the halogenated precursor using a ligand with more flexibility and larger bite angles. To this end, we set out to synthesize complexes of this type (Figure 5). While we have recently produced two such ligands, although no metal complexes have yet been made which contain either of these ligands.
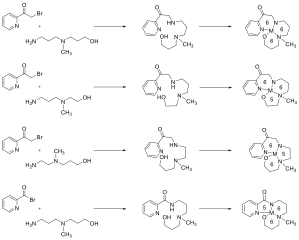
Figure 5. Synthesis of various ligand sizes to generate ring systems to reduce strain of the metal complex.
To this end, we also set out to streamline the synthesis of alternate ligands using solid phase peptide synthesis. This turned out to be a highly effective approach for generating alternate ligands like 9 (Figure 6). In fact, several hundred milligrams of this ligand was successfully produced from picolinic acid and subsequent additions of FMOC-beta-alanine to Rink amide resin. However, the procedure used to produce each of the metal complexes requires that the ligand be used in vast excess. In the interest of efficiency, we are in the process of generating a new procedure to generate metal complexes using less of this more precious ligand.

Figure 6. Successful synthesis of alternate peptide ligands, such as ligand 9 using FMOC-protected peptide synthesis on Rink-amide resin.
We then set out to examine the reactivity of each of the structurally characterized complexes in epoxide opening and epoxide formation reactions using a wide variety of conditions, by varying solvent, temperature, oxidant, substrate, etc. Since each of the metal complexes demonstrated low solubility in most organic solvents except methanol, most reactions required biphasic conditions to solubilize both the substrates and the catalyst, as well as any necessary oxidants. This is in sharp contrast to Jacobsen's epoxidation catalysts, which are quite soluble in relatively hydrophobic organic solvents like CH2Cl2. To address this solubility issue, peptide synthesis was used to generate novel ligands, with an eye toward adding hydrophobic groups to improve solubility, and eventually, stereocontrol. In addition to making more hydrophobic metal complexes, we also sought more polar substrates, like resveratrol, which could be used in a monophasic epoxidation reaction with the catalyst.
In lieu of a table of the hundreds of reactions we attempted to find a catalyst and conditions to epoxidize an alkene or open an epoxide we have included the following summary. We found no conditions where complexes 2, 3, 7, or 8 (20 mol %) would accelerate the opening of an epoxide of styrene oxide, stilbene oxide, or propylene oxide in MeOH, EtOH, iPrOH, ACN, DCM, EtOAc, Hexane, H20 or aqueous mixtures of these solvents at any temperature between 4oC and their respective boiling points. In addition, we found no conditions where complexes 2, 3, 7, or 8 (20 mol %) would accelerate the epoxidation of 1-octene, styrene, resveratrol, or stilbene in the aforementioned solvent mixtures at any temperature between 4oC and their respective boiling points.










