Reports: DNI653482-DNI6: Signature of Molecular Environment in Spectroscopy Measurement of Water and Aqueous Solutions Studied by Advanced ab Initio Methods
 printer friendly
printer friendlyPRF research progress report for the period of 09/01/15 – 08/31/16
During the PRF extension period from 09/01/15 to 08/31/16, we have mainly carried out the studies of two projects that are directly to the topic of the supported projects “Signature of molecular environment in spectroscopy measurement of water and aqueous solutions studied by advanced ab initio methods”. In the following, we have provided some details of the results and importance of the projects that we have finished during last year.
1. Accurate first-principles structures and energies of diversely bonded systems from an efficient density functional
One major difficulty in accurately predicting the spectra of liquid water and correctly assigning the spectra to its molecular environment is coming from the over-estimated H-bond strength and the corresponding over-structured liquid water structures in the conventional density functional theory (DFT) calculations. Within the ordinary DFT calculations, one atom or molecule binds to another through various types of bond, the strengths of which range from several meV to several eV. As a result, it is rather difficult to give accurate ab initio predictions based on local density approximation or the generalized gradient approximations. Although some computational methods can provide accurate descriptions of all bond types, those methods are not efficient enough for many studies (for example, large systems, ab initio molecular dynamics and high-throughput searches for functional materials). In the most recent work by collaborating with Prof. John P. Perdew who is a world-famous scientist in the area of density functional theory, we have shown that the recently developed non-empirical strongly constrained and appropriately normed (SCAN) meta-generalized gradient approximation (meta-GGA) within the density functional theory framework predicts accurate geometries and energies of diversely bonded molecules and materials (including covalent, metallic, ionic, hydrogen and van der Waals bonds). By capturing the intermediate range van der Waals interaction, we have shown that the SCAN functional is giving a rather accurate prediction of the H-bond structures of ices of various phases, and gas-phase water hexamer as shown in Fig. 1. The above accuracy in predicted the H-bonded materials could not be achieved by the conventional DFT approaches such as PBE functional as the most used functional in predicting the liquid water structures. Our results have been published in one of the high impact journals, Nature Chemistry, 8, 831 (2016). The ACS PRF supported is also acknowledged in the published for the research parts contributed by my group.
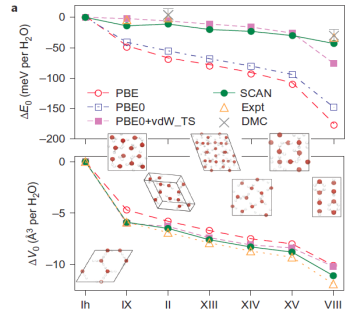
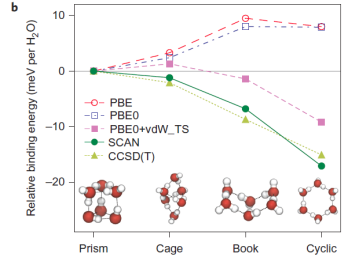
Fig 1: SCAN captures the intermediate-range, many-body van der Waals interactions necessary for a quantitative description of various ices and gas-phase water hexamers. a, The relative sublimation energy ΔE0 and equilibrium volume change ΔV0 per water molecule of seven hydrogen-ordered ice phases with respect to the ground-state ice Ih illustrate that SCAN is the only functional tested that predicts the relative stability of ice phases in quantitative agreement with experimental results. The sublimation energy (E0) is the energy needed to break an ice into isolated water molecules. b, The relative binding energy per water molecule of four low-energy water hexamers similarly illustrates that SCAN is the only semi-local density functional approximation that predicts the known energetic ordering of these clusters, evidenced by the agreement between SCAN and CCSD(T). The zero-point energy effects have been removed from the experimental results for ice. Data points (and error bars) computed by methods other than SCAN are from the ice phases and water hexamers, respectively. Lines are guides to the eye.
2. Signature of H-bond environment of liquid water in X-ray emission spectra by ab initio methods
By using ab initio methods, a student, my postdoc, and I have recently studied the relation between the hydrogen bond and the x-ray emission spectroscopy (XES) of liquid water. The existence of hydrogen bond in liquid water makes its XES different from that of gas water. However, the effect of the H-bond strength and H-bond network on the XES spectra of liquid water has not been studied in details so far. In this project, we carried out systematic studies to address the questions: how hydrogen bond in liquid water can affect the XES by using ab initio methods. By considering the core-hole induced dynamics, we observe the OH bond dissociation which is very sensitive to the donating hydrogen bond as shown in Fig. 2. The dissociation of OH bond can further affect the molecular orbitals and DOS of liquid water. Especially, the bonding orbital 1b2 and 3a1 are very sensitive to the OH bond dissociation. Therefore, the hydrogen bond can affect the XES through the core-hole induced dynamics. Our results give an interpretation of the XES based on the H-bond structure in condensed phase. Therefore, we conclude that the OH bond dissociation has a great effect on the molecular orbitals and DOS of liquid water. Especially, the peaks of bonding orbital 1b2 and 3a1 in XES are very sensitive to the donating hydrogen bonds in liquid water. We have finished most of the calculations and analysis. The manuscript is under preparation to be submitted soon.
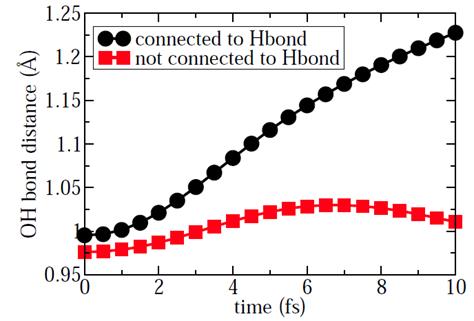
Fig. 2 The OH bond dissociation represented by the OH bonds distance for the X-ray excited water molecules as a function of time.










