Reports: DNI555581-DNI5: Descriptor-Based Molecular Design of Metal-Organic Frameworks for Catalytic C-H Activation
 printer friendly
printer friendlyThe central theme of the project is to develop design concepts for metal-organic frameworks catalysts discovery. We start by summarizing the two main accomplishments of the project.
1. We have mapped out selective CH4 oxidation reaction kinetics using MOF-74 to molecule-surface interactions, to be specific, the *OH binding energies.
First-principles-based kinetic models have the ability to connect properties at the molecular level, e.g., binding energies of surface intermediates and activation energies of elementary steps, to measurable quantities such as reactivity, selectivity, and stability. Microkinetic modeling as a mean-field approach has been used routinely in catalysis research and become an invaluable tool for understanding how existing catalysts work and predicting new ones with improved performance. Within the current project, ab initio electronic structure calculations have been conducted to calculate the thermodynamics and kinetic parameters of elementary steps in selective methane oxidation to methanol with O2 on redox-active MOFs. It is well known that paraffinic hydrocarbon can be adsorbed on a strong Lewis acid sites [1] that are abundant on the MOF-74 and HKUST-1 family MOFs targeted in the current project. We observed a strong linear correlation between adsorption energies as shown in Fig. 1(a) for a series of MOF-74 (see Fig. 1(b)). The slopes of linear scaling relations on MOFs for OH vs. O and OH vs. OCH3 determined by the valence of molecules are 2 and 1, respectively, is strikingly similar with that on metal surfaces [2–4].
Thanks to the scaling relations between adsorption energies of different intermediates the binding energy of a single intermediate or two are often sufficient to describe the trend of catalytic activity, greatly simplifying the picture for searching new catalysts. We expected that the reaction rates from kinetic modeling exhibit volcano-shaped relationships with respect to the chemical descriptor, i.e., the binding energy of a surface intermediate such as OH. The optimal MOFs catalyst for methane partial oxidation should be active enough to break O-O bonds, while also not too reactive that methoxyl (CH3O) and other intermediates will not easily desorb to release the active sites.
2. We have identified key electronic factors governing molecule-surface interactions on relevant organometallic complexes, homogeneous analogue of MOFs.
Design of catalytic materials with optimized performance relies on a fundamental understanding of the electronic structure of an ensemble of surface atoms and its relationship with energetics of molecule-surface interactions. For that matter, identification of simple reactivity descriptors from complex electronic properties of materials becomes tremendously important. Such a simplification often leads to key concepts that can provide guidance in tailoring the geometry and composition of surface atoms for desired properties.
In this objective, we identify physical factors governing the intrinsic reactivity of grafted Ir-based molecular catalysts on MOFs for O-O bond activation for electrochemical water oxidation. We note that this type of hybrid MOFs catalysts is highly relevant for C-H bond activation. We took our initial efforts for O-O bond activation because the mechanism of O-O bond activation is well established in electrocatalysis community. We will extend this understanding to the C-H bond activation as outlined in our proposal. Since the stability of HO*, *OH, O*, and *OOH intermediates are linearly scaled with each other, so we use Ir-oxo (O*) intermediate as the reactivity descriptor. The high-spin Ir(V)=O intermediate is a strong electrophile that can abstract a hydrogen atom from a water and form the Ir(III)-OOH intermediate. The motivation for this Task is to identify underlying electronic factors that govern the thermodynamic stability of the O* intermediate (the descriptor) and eventually the kinetics of reactions. Since the binding energy of reaction intermediates is mainly governed by local electronic properties of molecular catalysts, we will use the corresponding Cp*Ir complexes as the model system as illustrated in Fig. 2. We found that the molecular orbital energies are playing an important role in determining kinetics of charge transfer reactions. We will use the corresponding electronic structure of molecular complexes, such as the LUMO orbital energies of reaction intermediates as the descriptor. We observed that the Ir-oxo LUMO state energy has a strong correlation to the reaction energy from the Ir-oxo to Ir-OOH step. We used the Cp*Ir with bpy, ppy, and their analogues as the coordinating ligands on the hybrid organometallics/MOFs catalysts. We note that a similar approach has been used recently for Fe-based complexes for C-H bond activation [5]. In this effort, we are going to explore a broad range of Cp*Ir-based molecular complexes to develop predictive structure-activity relationships.
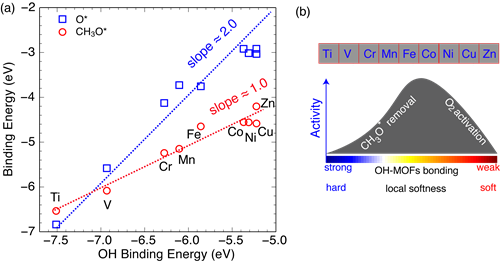
Fig. 1 (a) Scaling relations of binding energies of surface intermediates on the MOF-74 family, (b) Schematic of the methanol production rate as a function of reactivity descriptors
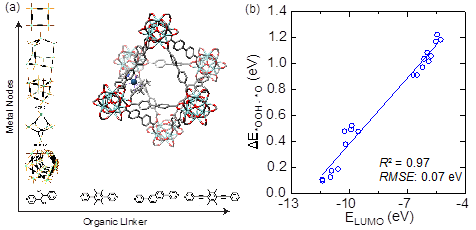
Fig. 2 (a) Organometallics-MOFs model systems with varying metal nodes and organic linkers, (b) Correlation between the energy of the LUMO state of the anchored Ir-oxo complex on UiO-67 and the reaction energy of the O-O bond formation step with the OPBE functional.
1. References
[1] Y. Moro-oka, Appl. Catal. Gen. 181, 323 (1999).
[2] F. Abild-Pedersen, J. Greeley, F. Studt, J. Rossmeisl, T. R. Munter, P. G. Moses, E. Skulason, T. Bligaard, and J. K. Norskov, Phys. Rev. Lett. 99, 16105 (2007).
[3] E. M. Fernández, P. G. Moses, A. Toftelund, H. A. Hansen, J. I. Martínez, F. Abild-Pedersen, J. Kleis, B. Hinnemann, J. Rossmeisl, T. Bligaard, and J. K. Nørskov, Angew. Chem. Int. Ed. 47, 4683 (2008).
[4] G. Jones, F. Studt, F. Abild-Pedersen, J. K. Nørskov, and T. Bligaard, Chem. Eng. Sci. 66, 6318 (2011).
[5] P. C. Andrikopoulos, C. Michel, S. Chouzier, and P. Sautet, ACS Catal. 5, 2490 (2015).










