Reports: ND354739-ND3: New Ligands for Catalysis Through Elaboration of the Phosphatriptycene Framework
 printer friendly
printer friendly
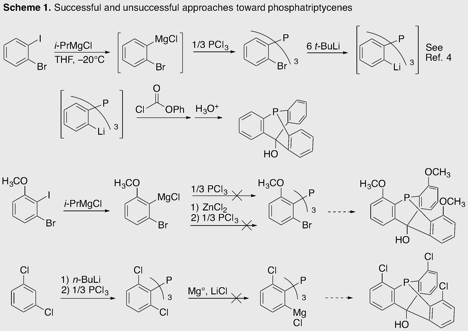
At the beginning of this reporting period, we had obtained a simple 10-hydroxy-9-phosphatriptycene by modification of a published route to 10-sila-9-phosphatriptycenes (Scheme 1).4 We sought a route to phosphatriptycenes substituted in the positions flanking the phosphorus donor, as originally proposed. These proved elusive. While 2-(bromophenyl)magnesium chloride reacts with phosphorus trichloride to form tris(2-bromophenyl)phosphine, 2-bromo-6-methoxyphenylmagnesium chloride undergoes significant decomposition through benzyne formation. The corresponding zinc reagent, far less prone to benzyne formation, proved too mild to replace all three chlorides on phosphorus. A method for selective conversion of 1,3-dichlorobenzenes to 3-chlorophenylmagnesium chlorides,5 which we reproduced successfully, gave no reaction when the readily obtainable tris(2,6-dichlorophenyl)phosphine6 was used as substrate. Both the trimethoxy- and trichloro-substituted phosphatriptycenes were to be used in the synthesis of more elaborate architectures. We concluded that steric and entropic costs make their synthesis difficult.
Shifting focus slightly, we reasoned that substituents meta to the phosphorus might be more readily incorporated. We could imagine building a bowl around the ligand that would engender low coordination numbers, yet present relatively little hindrance close to a bound metal center. Initially, we envisioned tert-butyl substituents in the meta-positions. For synthetic convenience, and to achieve potentially a better pi-acceptor than the parent phosphatriptycene, we pursued the 10-aza-9-phosphatriptycene skeleton, for which a triarylamine appeared a reasonable synthon.
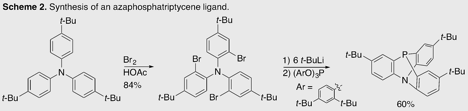
The solid 4,4'-di-tert-butyldiphenylamine is readily available at reasonable cost (25g for ~$25). Palladium-catalyzed arylation using 4-tert-butylbromobenzene according to a known method7 affords 4,4',4''-tri-tert_-butyltriphenylamine in good yield on multigram scale. Bromination of this amine forms 2,2',2''-tribromo-4,4',4''-tri-tert_-butyltriphenylamine cleanly. Lithiation, followed by quenching with the crystalline tris(2,4-di-tert-butylphenyl) phosphite affords the corresponding azaphosphatriptycene in good yield. This sequence is similar to that used by Hellwinkel,3 but the earlier preparation, in its authors' hands and ours, afforded poor and irreproducible yields. The method outlined here affords the ligand reproducibly, on gram scale, with no chromatography required.
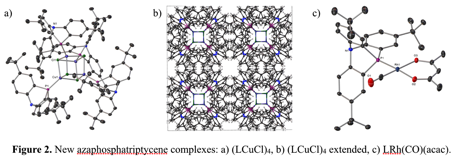
We have so far isolated two metal complexes of this ligand: the copper(I) chloride, which crystallizes as a tetramer with a heterocubane structure (Figure 2a, 2b), and the (azaphosphatriptycene)rhodium(I) (carbonyl)(acetylacetonate) (Figure 2c). Sterically, this ligand happens to be surprisingly similar to triphenylphosphine itself; constraining the phenyl groups to a triptycene framework results in a much smaller ligand, and then the tert-butyl substituents restore some steric demand.
In preliminary results, the rhodium complex appears to be, as hoped, a highly active catalyst for alkene hydroformylation. We plan to confirm these early findings, and in future work, if possible, to explore the incorporation of far larger substituents at the periphery of the ligand.
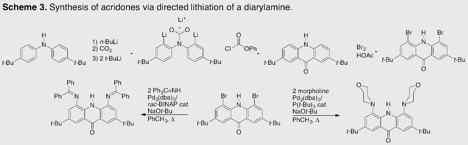
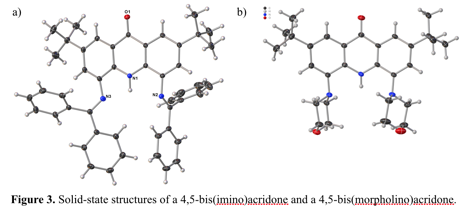
In parallel to this work, we have continued to explore the chemistry of 4,5-disubstituted acridones. The synthesis of the 4,5-dibromo ligand is recapped in Scheme 3. Reasoning that a pincer ligand should facilitate binding, we appended benzophenone imine moieties to the acridone, but found that the phenyl rings proximal to the acridone backbone are unable to lie flat in the desired arrangement (Figure 3a); thus, the imine nitrogens point in the wrong direction to relieve strain! We next incorporated the morpholino group, which did facilitate metalation (Figure 3b). Although solid-state structures remain elusive, we have evidence (UV-visible, infrared, and mass spectrometric) for the formation of both chlorocobalt(II) and chlorocopper(II) complexes of the acridone anion. A promising feature of this ligand is its conversion from an anion to a neutral donor by protonation at the peripheral oxygen, which should greatly affect the redox potential of the supported metal center. We look forward to exploring this feature through electrochemical investigation, and hope to employ this principle in electrocatalytic small molecule reductions.
References
1. Jongsma, C.; De Kleijn, J. P.; Bickelhaupt, F. Tetrahedron 1974, 30, 3465Ð3469.
2. Huszthy, P.; Vermes, B.; B‡thori, N.; Czugler, M. Tetrahedron 2003, 59, 9371Ð9377.
3. Hellwinkel, D.; Schenk, W.; Blaicher, W. Chem. Ber. 1978, 111, 1798–1814.
4. Iwai, T.; Konishi, S.; Miyazaki, T.; Kawamorita, S.; Yokokawa, N.; Ohmiya, H.; Sawamura, M. ACS Catal. 2015, 5, 7254–7264.
5. Dunst, C.; Knochel, P. Synlett 2011, 2064–2068.
6. Palau, C.; Berchadsky, Y.; Chalier, F.; Finet, J.-P.; Gronchi, G.; Tordo, P. J. Phys. Chem. 1995, 99, 158–163.
7. Li, M.-H.; Hsu, C.-W.; Shen, P.-S.; Cheng, H.-M.; Chi, Y.; Chen, P.; Guo, T.-F. Chem. Commun. 2015, 51, 15518–15521.










