Reports: ND154452-ND1: Metal-Free Functionalization of Aromatic Rings - Remarkably Mild Carbon--Heteroatom and Carbon-Carbon Bond Formations, and Dehydrogenative Cross-Coupling Reactions
 printer friendly
printer friendlyWe are exploring transformations that leverage the nitrogen functionality of aniline to enable selective reactivity on the aromatic ring. Transient oxidation of the amino group affords us the ability to introduce carbon–heteroatom and carbon–carbon bonds under exceptionally mild reaction conditions. The oxidation event exploits the typical liability experienced with anilines and enables controllable, metal-free, and environmentally friendly reactions. Subsequent O-acylation temporarily transforms the aniline ring into an electron deficient species allowing selective and controlled introduction of new carbon–heteroatom and carbon–carbon bonds on the aromatic ring, while retaining the classical regiochemistry associated with electrophilic aromatic substitution.
The original goals of this proposal were to harness the elevated reactivity of N,N-dialkylaniline-N-oxides to: (1) complete the development of new carbon–heteroatom bond formations on aromatic rings, (2) to develop direct carbon–carbon bond formations on aromatic rings, (3) to develop metal-free dehydrogenative cross-coupling reactions of N,N-dialkylanilines, and (4) to develop the direct trifluoromethylation of N,N-dialkylanilines.
In the first year of support, we made considerable progress in the manipulation of the aniline-N-oxide platform taking advantage of [3,3]-sigmatropic rearrangements. We described success in goals 1 and 2 with protocols for the C-alkylation of N,N-dimethylaniline-N-oxides, as well for the synthesis of amino alcohols, amino triflates and tosylates (Figure 1).

Figure 1. Recently described bond formations on the aniline N-oxide platform.
In each protocol, an acylation event at oxygen that gives a p–system is believed to facilitate an asynchronous [3,3]-sigmatropic rearrangement to excise the weak nitrogen-oxygen bond and form the new carbon–heteroatom or carbon–carbon bond of interest. Ongoing computational studies support this conclusion.
We demonstrated the direct formation of carbon–carbon bonds utilizing ethyl malonyl chloride, cyanoacetyl chloride, and diketene as acylating agents that could present a carbon–carbon p–system for the subsequent [3,3]-sigmatropic rearrangement. A single literature reference reported ketene as a viable participant in reactions with aniline-N-oxides, albeit with several side products attributed to multiple mechanistic pathways that might excise the weak nitrogen-oxygen bond. We encountered no such difficulties.
One major liability that we have encountered at the aniline-N-oxide oxidation level is the simple Polonovski-type elimination reaction that may occur prior to group transfer, which typically produces a small amount of dealkylated aniline products. However, during our attempts to generate polyfunctionalized anilines by successive oxidation cycles, we found that substrates that contain a nucleophilic function ortho to the aniline can undergo a Mannich-type reaction in which the previously unproductive iminium ion is captured to give substituted indolines products. We took advantage of this observation and developed a protocol for the convenient synthesis of a variety of indolines with opportunities for further elaboration. The reaction appears to be general and tolerant of both electron-withdrawing and electron-donating groups on the aromatic ring (Figure 1, middle). We found both the ethyl esters and nitriles to be competent participants in the Polonovski-Mannich cyclization reaction.
In the second year of support, we have greatly expanded the landscape of acylating agents that can present a carbon–carbon p–system (or equivalent) for a subsequent [3,3]-sigmatropic rearrangement. We treated N,N-dimethylaniline-N-oxides under Swern conditions (chlorodimethylsulfonium ion), and upon warming with triethylamine as base, we observed formation of the corresponding sulfoxide products (Figure 2a). These sulfoxides will allow access to a host of useful synthetic building blocks including sulfones, aldehydes, and thioethers (Figure 2b). Though this acylation event at oxygen does not give the same p–system as other electrophiles, we still favor an asynchronous [3,3]-sigmatropic rearrangement to give the products. Ongoing computational studies support this conclusion. Consistent with this work, treatment of N,N-dimethylaniline-N-oxides with nitroethylene at low temperature followed by warming with triethylamine gives highly versatile nitroalcohol alkylation products (Figure 2a) that allow access to another large variety of synthetic building blocks including aminoalcohols, unnatural aminoacids, and heterocyclic scaffolds (Figure 2b).
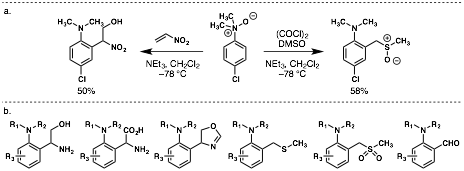
Figure 2. a. New rearrangement products facilitated by aniline-N-oxides. b. New synthetic building blocks available from a single precursor.
In the second year of support, we have also begun to explore a new manifold in aniline-N-oxide reactivity that is facilitated by acylation events that are not followed by a [3,3]-sigmatropic rearrangement. In these reactions, treatment of N,N-dimethylaniline-N-oxides with di-tert-butyl dicarbonate gives activated tert-butyl carbonates in which the nitrogen atom now bears a leaving group. These carbonates are long-lived, do not transfer the acyl group to the aromatic ring even after prolonged stirring at room temperature (and elevated temperatures), and is characterizable by NMR spectroscopy (Figure 3). Upon exposure of these species to exogenous nucleophiles, we observed formal metal-free dehydrogenative cross coupling reactions to give functionalized biaryl compounds and alkylated anilines (Figure 4a). Thus far, we have experienced success with indoles to give biaryl compounds.

Figure 3. Activated aniline-N-oxides that do not transfer the acylated group.

Figure 4. a. A metal-free dehydrogenative cross-coupling reaction. b. The proposed mechanism of this transformation.
At this stage in our work we favor a mechanism in which the nucleophile engages the aniline in a substitution reaction analogous to an SN' type process in which the carbonate is lost and aromaticity is briefly disrupted (Figure 4b). A series of proton transfers restores aromaticity (of the aniline and in some cases, the electron rich aromatic nucleophile). More detailed mechanistic studies are ongoing, and this chemistry will be described in the near future, however we have validated the feasibility of aims 3 and 4 of the proposed research as we enter our last year of support.










