Reports: ND155606-ND1: Synthesis of Cyclacenes: Strained Macrocyclic Hydrocarbons with Unique Bonding
 printer friendly
printer friendlyEfforts towards achieving our first aim, the design and assembly of curved-strained macrocyclic precursors to a cyclacene, began with investigating the installation a cyclopentadienone to key intermediate 1 (Scheme 1). Graduate student Sarah Wegwerth carried out a model study (Scheme 2) using benzonorbordiene 8. Surprisingly, the disubstituted alkene of the norborndiene ring reacted sluggishly with OsO4. Under the optimized conditions,(1 week at RT), only a 60% conversion of 8 to the diol 9 was observed. At elevated temperatures (40 °C) a by-product, presumably the dihydroxylation of the bridgehead alkene, was also observed. A Swern oxidation of 9 smoothly provided dione 10. In the presence of light, 10 rearranged to tetracyclic oxo-enol 12. Attempts to advance 10, in the dark, revealed that it is unstable under basic conditions at elevated temperatures. Due to the sluggish first step and instability of intermediate 10, other routes were investigated.
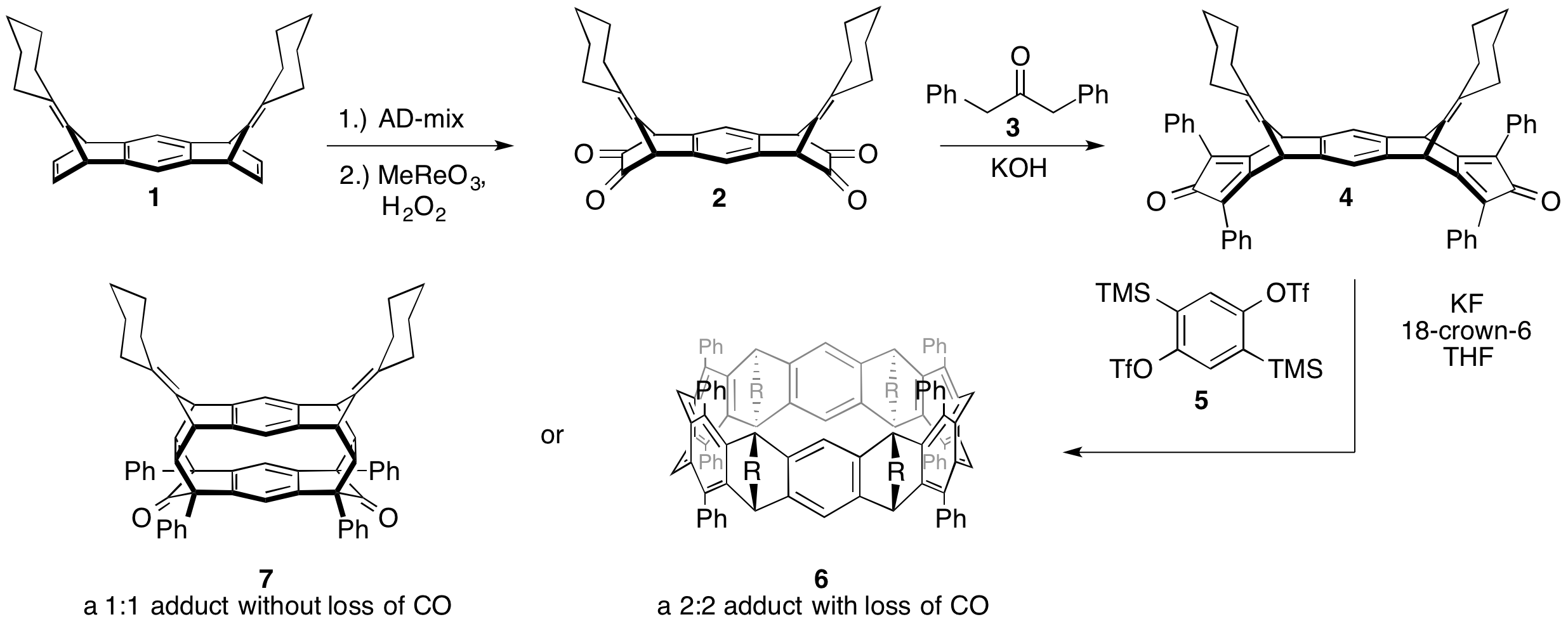
Scheme 1: Proposed elaboration of 1 to the cyclacene precursors bis(cyclopentadienone) 4
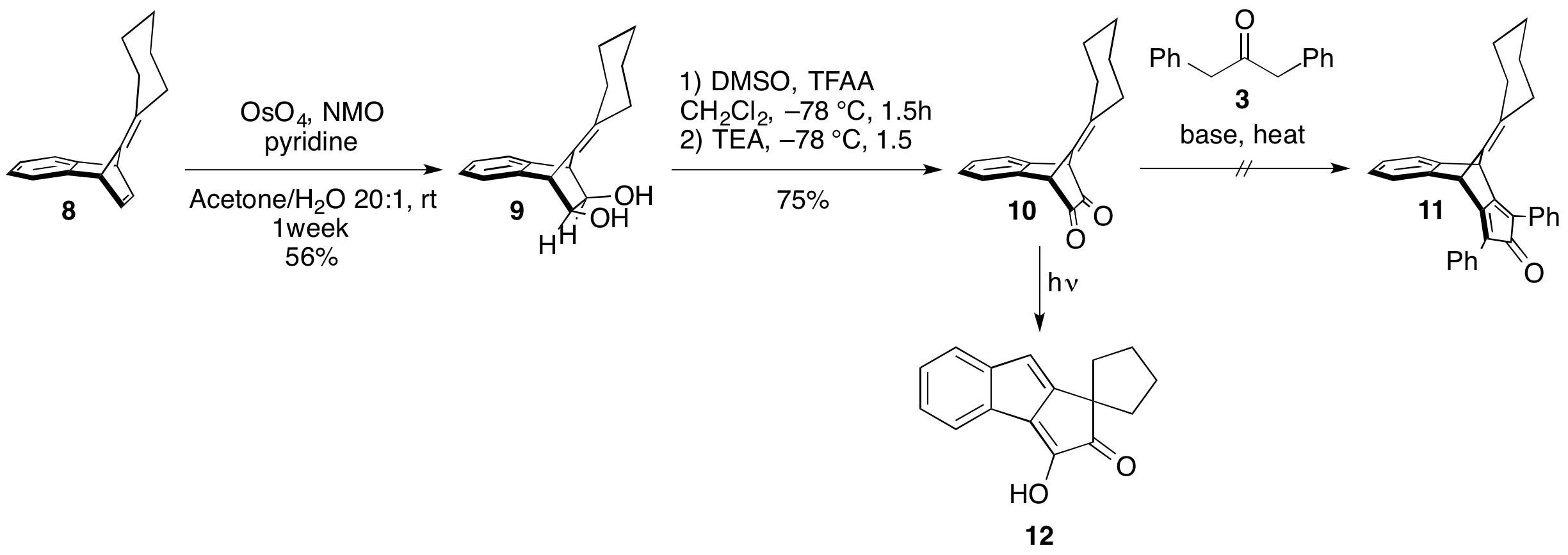
Scheme 2: Model study of the installation of the cyclopentadienone moiety
Initially, we proposed to convert the norbornene alkene of 1 into an enamine to participate in an inverse electron demand Diels–Alder reaction with tetrazene 15 (Scheme 3). The resulting product would be bisdieneophile 16. Participation of 16 in Diels–Alder reactions with 14 could yield the desired macrocycle. Graduate student Sarah Wegwerth investigated formation of the enamine, using model benzonorbornodiene 8, with minimal success. Reexamination of the literature revealed that enamine formation was not necessary. Upon treatment of benzonorbornodienes with tetrazenes, such as 17, isobenzofulvenes (18, Scheme 4) form. These isobenzofulvenes can be trapped with quinones. If 2,5-dibromoquinone is used, then dehydrohalogenation can re-form the quinone.
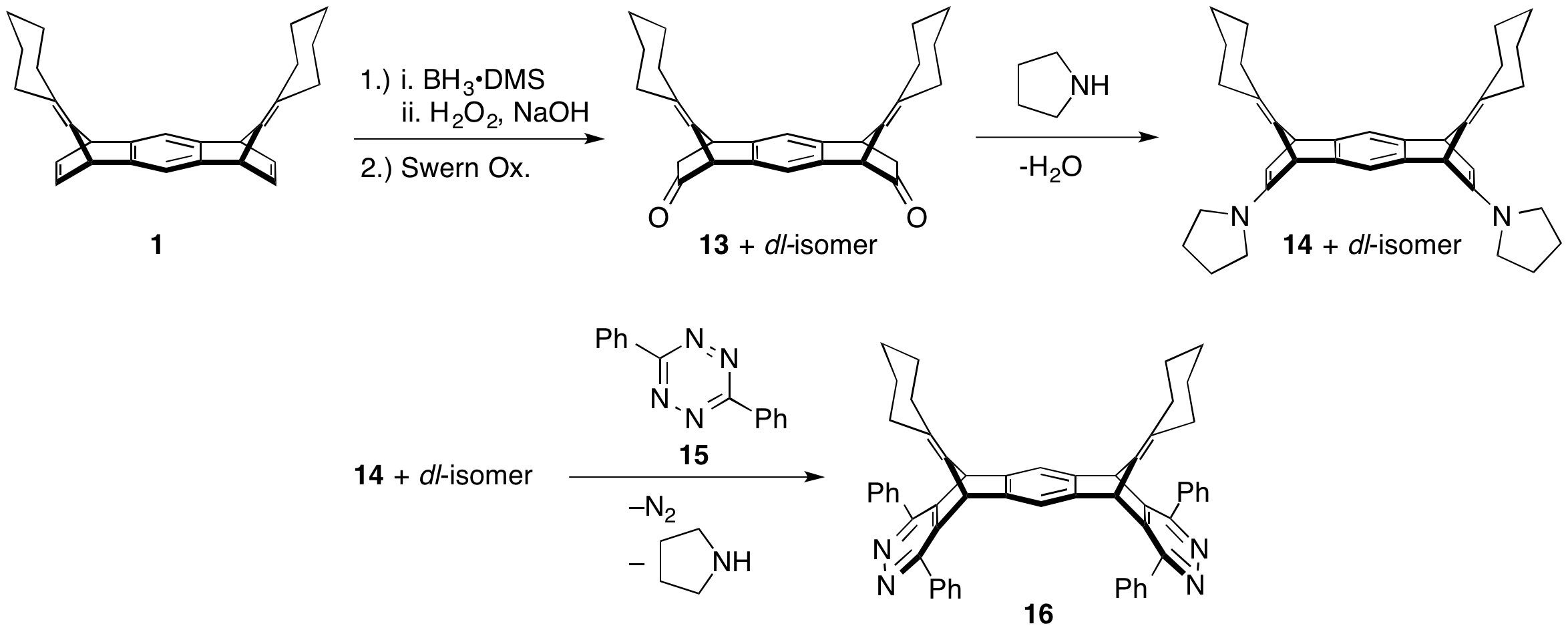
Scheme 3: Proposed conversion of 1 to macrocyclic precursors
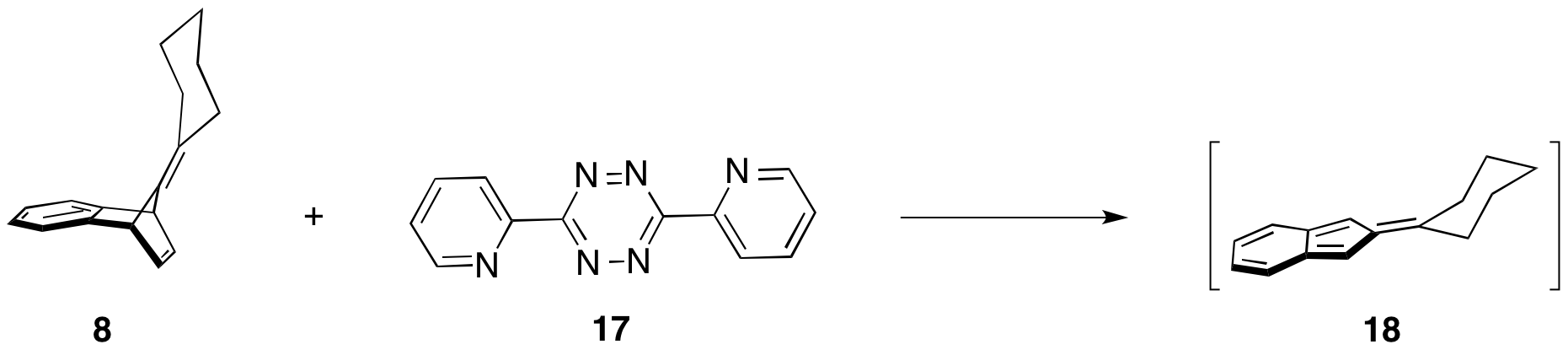
Scheme 4: Formation of isobenzofulvene 18 from 8.
To probe the utility of this chemistry, Wegwerth performed a model study. Based on NMR studies, the reaction is complete within a half an hour, cleanly producing a 1:6 mixture of the exo and endo dihydroquinones 20 and 21. This mixture was inconsequential as upon passing through a basic alumina column both were converted to bromoquinone 22 in 79% over the two steps. Subjecting quinone 22 to a second equivalent of the isobenzofulvene formed from 8 and 19, and subsequent treatment with basic alumina yielded a 1:1.5 mixture of the syn and anti products 23 and 24 with a combined yield of 99% over two steps!
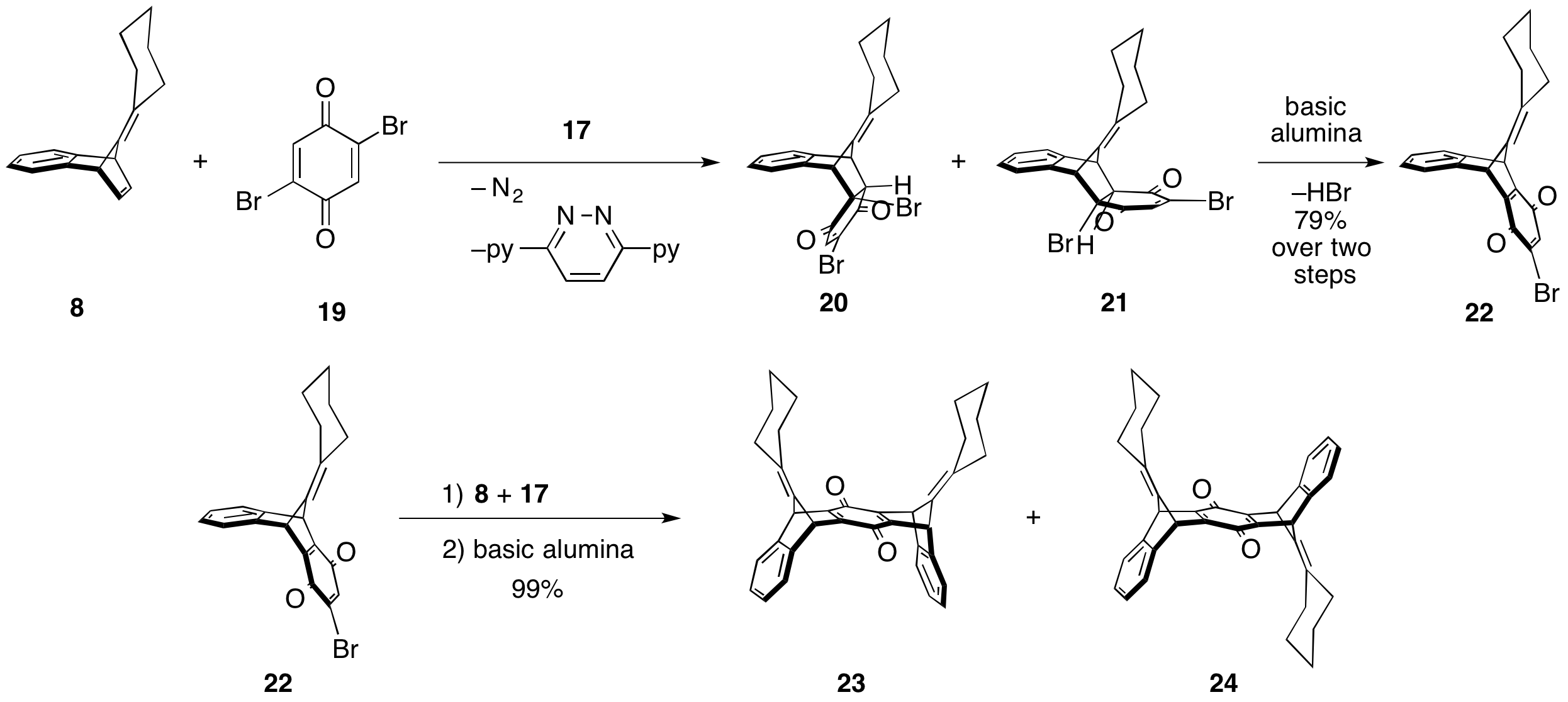
Scheme 5: Model study investigating formation of isobenzofulvene and trapping with 2,5-dibromoquinone.
The success of the model study encouraged us to continue pursuing isobenzofulvene as our diene and quinones as the dienophiles. This new strategy was attractive for a couple of reasons. First, it advances both the syn and anti isomers of key intermediate 1, as bridgehead stereochemistry is ablated in the formation of the isobenzofulvene. Second, the quinones allow a handle for late-stage installation of edge group. This will allow us to more efficiently investigate aim 2 (identify substituents that provide kinetic stability to the cyclacene core).
Wegwerth's approach to macrocycle focused on advancing the bis(norbornodiene)s 1 and 25. The initial strategy was to install the quinone functionalities on both sides of 1 and 25 in one reaction. This however resulted in essentially only the anti product 26 which could not be carried forward to macrocycle. Stepwise addition of the quinone groups yielded about a 1:1.6 mixture of the syn to anti products 29 and 30 and their respective bromine constitutional isomers. Preliminary work on elongating the syn-isomers of 29 has successfully added an equivalent of 1/25.
Attempts at closing an 8-benzenoid-ring macrocycle have been inconclusive. Currently the hypothesis is that a 12-ring macrocycle, targeted through adding another equivalent of 1/25 and 19, will be less strained and kinetically accessible. Work was also done to dimerize 27/28, however this reaction proceeded with low conversion. Again, we hypothesized that the 8-ring macrocycle precursor does not allow for the diene and dienophile to come close enough together for the final Diels–Alder reaction.
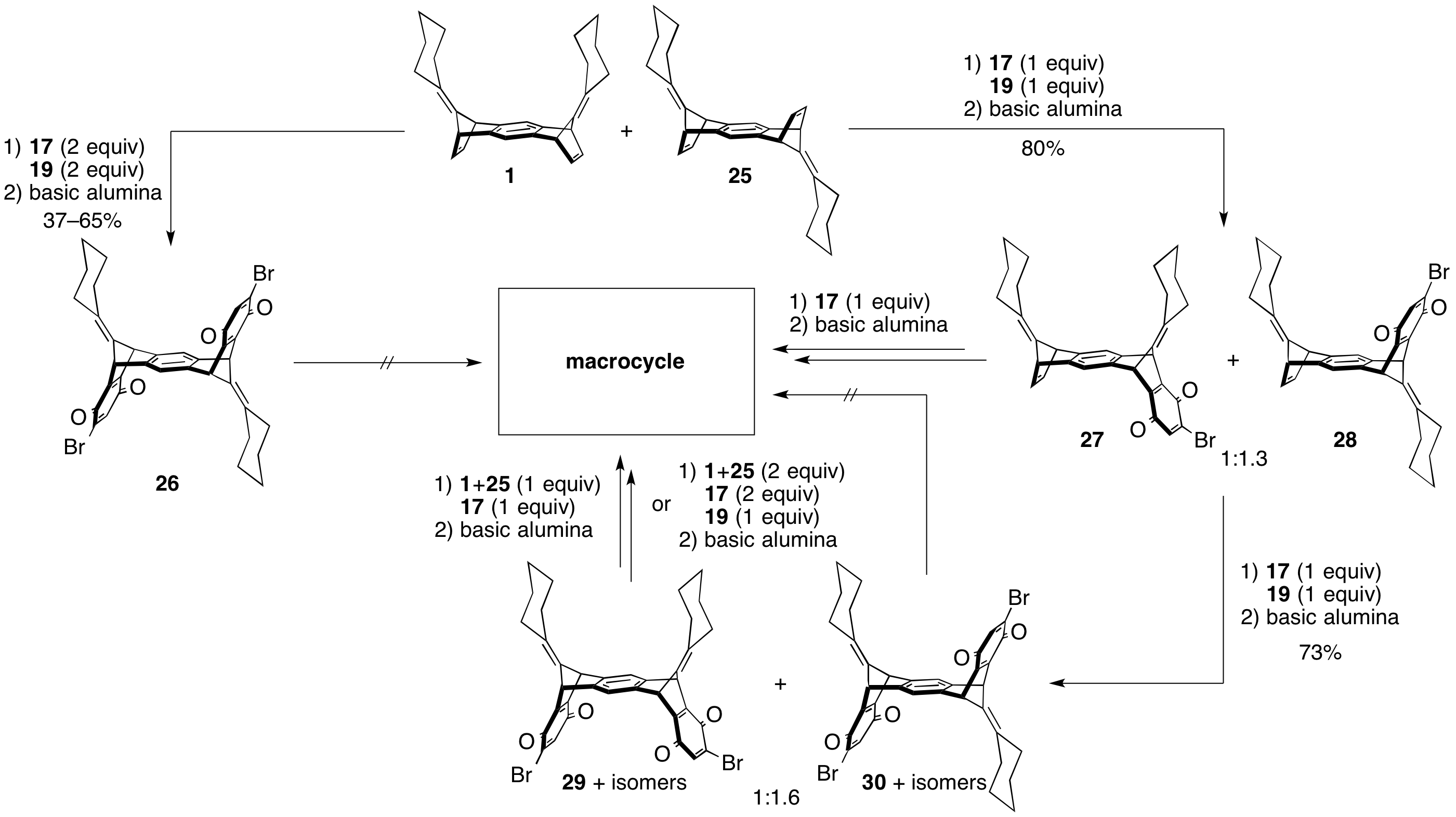
Scheme 6: Work done to advance 1/25 towards macrocycle.
Graduate students Zhuoran Zhang and Lafe Purvis started the synthesis by a benzyne Diels–Alder cycloaddition between the disilyltriflate (benzyne precursor, 5) and cyclohexylfulvene (diene, 31) (Scheme 7). The reaction smoothly afforded the desired mono-bridge cycloadduct (32) under the control of reaction temperature and cesium fluoride equivalence. Next, the mono-bridge cycloadduct (32) was transformed into bromodione (33) in a one-pot reaction. The mono-bridge cycloadduct was first treated with dipyridyltetrazine (17) to form the isobenzofulvene intermediate (34), which was quickly trapped by dibromobenzoquinone (19) generating the dibromide (35). Dehydrobromination was then achieved by treating the solution with basic alumina during the workup. We note that this one-pot transformation gives the bromodione (33) as a 1:1 mixture of the cis/trans regioisomers. Utilizing the same methodology, the bromodione (33) was further extended to the double-bridge cycloadduct 36. Theoretically, this step could generate sixteen stereoisomers based on multiple stereogenic centers. However, the number of possible stereoisomers decreases to four upon dehydrobromination. By careful NMR and XRD analysis, the double-bridge dione (38) was identified as a 5:1 mixture favoring the syn configuration of the two cyclohexylidine units, regardless of the inconsequential regioisomers resulting from the TMS groups. The desired syn-isomers were purified by crystallization.
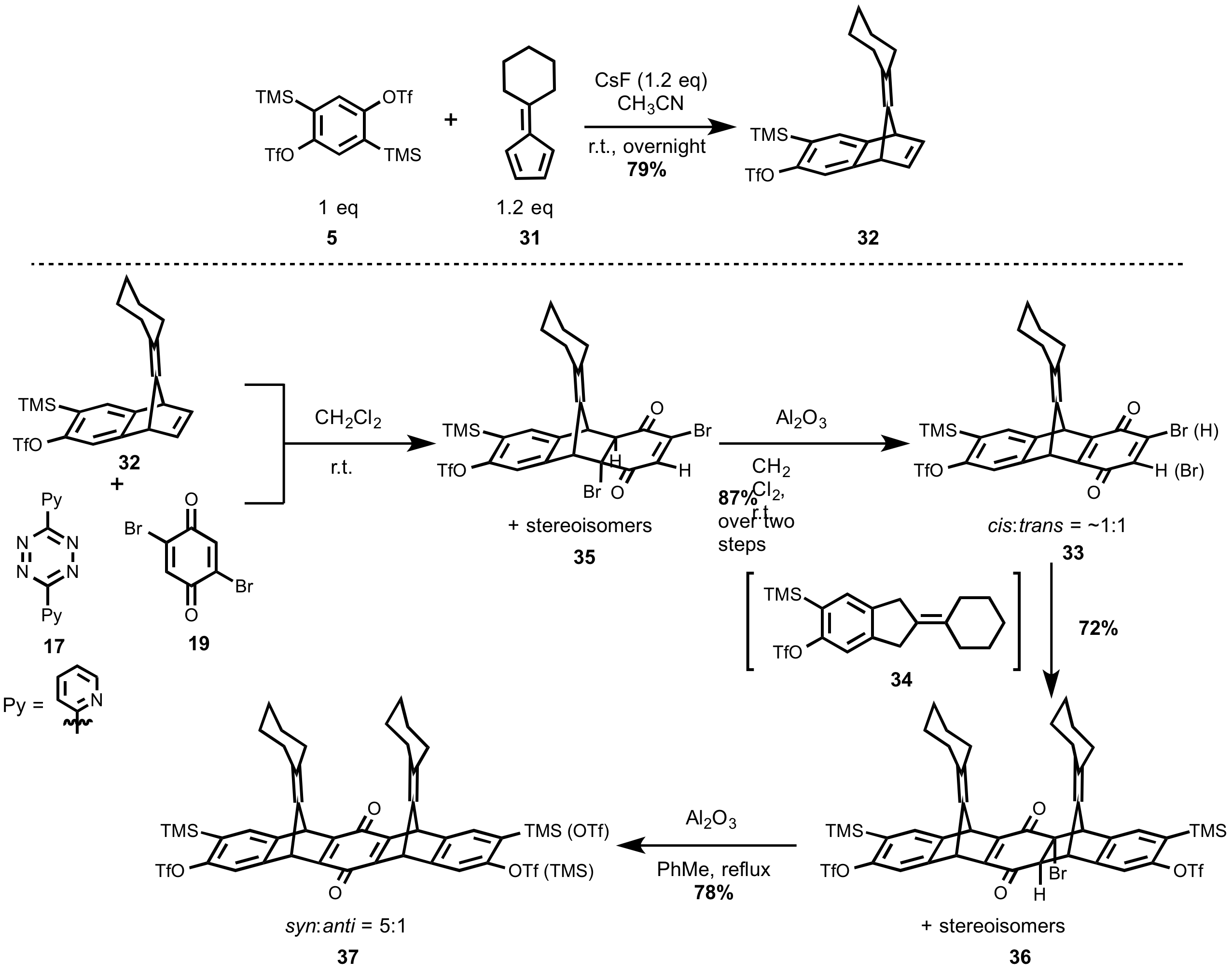
Scheme 7. Synthesis of the syn-isomer 37.
To explain the stereoselectivity favoring the syn-isomers, Zhuoran proposed a model to compare the steric encumbrance during the Diels–Alder reaction between the bromodione (33) and isobenzofulvene (34). Noticing that in the transition state shown at the top, the diene approaches from the exo face of the dienophile in an exo Diels–Alder reaction manner (Figure 1). This approach avoids any steric encumbrance involving the bulky silyl triflate group, and is regarded as the most favorite transition state.
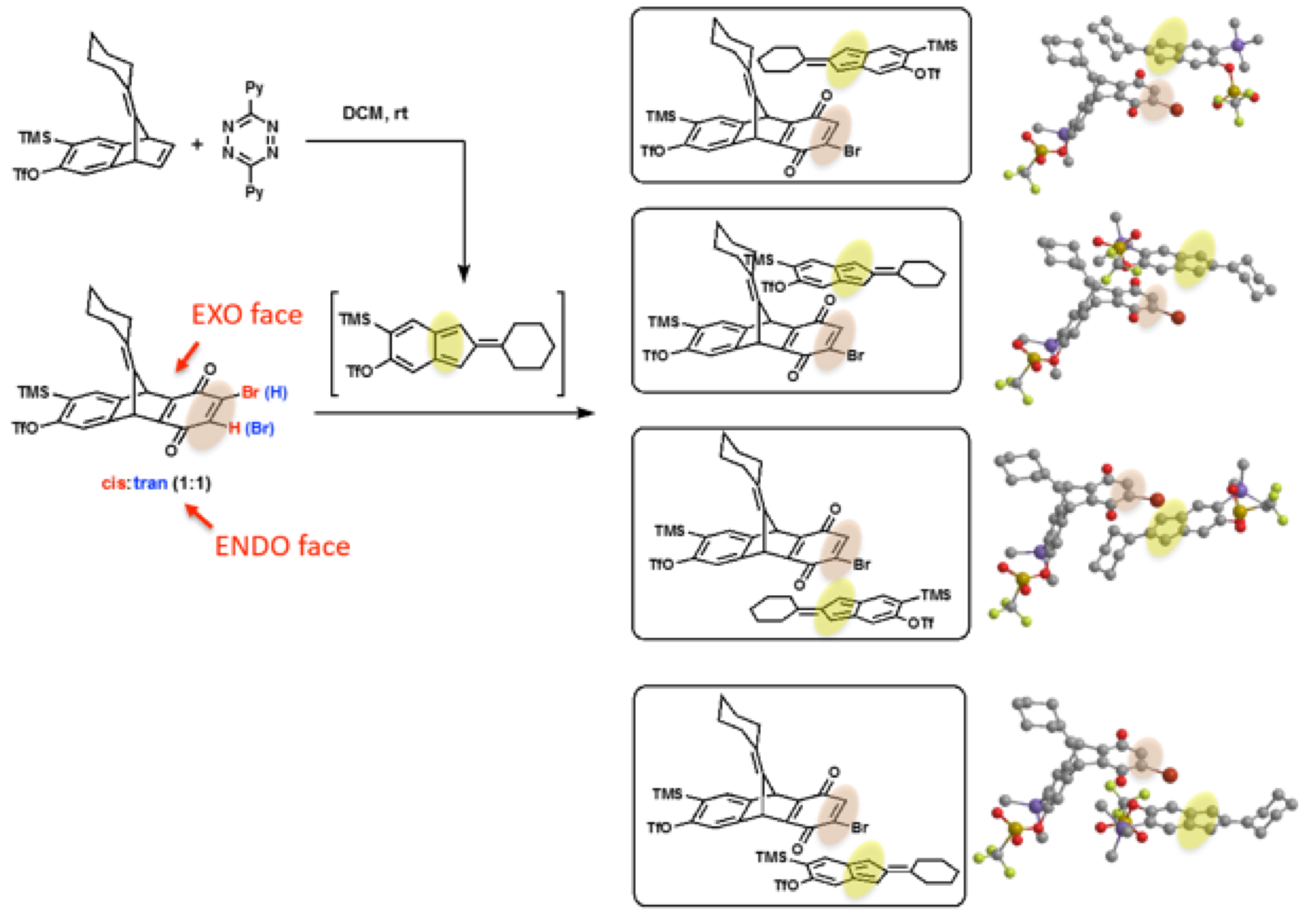
Figure 1. Steric analysis of Diels–Alder cycloaddition between 33 and 34.
Future work focuses on constructing the macrocycle via syn-isomer 37. Studies of late stage modification via quinone functionalization and modeling the oxidation of the cyclohexylidine are also underway.










