Reports: DNI354225-DNI3: Fundamental Studies toward the Copolymerization of Ethylene with Carbon Dioxide: Bimetallic Group 10/Main Group Catalyst Synthesis and Reactivity
 printer friendly
printer friendlyResearch in the Tonks lab is focused on synthesizing bimetallic catalyst systems to copolymerize carbon dioxide with ethylene into biodegradable polyesters. CO2/C2H4 copolymerization is thermodynamically disfavored below a 1 : 2.37 ratio of CO2 : C2H4. As a result, hypothetical CO2/C2H4 copolymerizations with late transition metal catalysts proceeding through consecutive insertions of CO2 and C2H4 are kinetically inaccessible because there is a high-energy barrier en route to endothermic CO2 insertion relative to essentially barrierless exothermic C2H4 insertion.

Figure 1. Bimetallic chain transfer strategy for CO2 and ethylene copolymerization.
Our approach has been to utilize recent advances in chain transfer polymerization to in situ functionalize growing metal-polyethyl chains with CO2 (Figure 1). This will be accomplished by designing polymerization systems capable of polymeryl chain transfer equilibrium between two different metal centers: one transition metal center that is capable of ethylene polymerization, and a second metal center uniquely capable of alkyl (polymeryl) group insertion into CO2 to form a carboxylate. The resulting polymeryl carboxylates can then rebound to the ethylene polymerization metal via Wacker-type ethylene carboxylation, closing the catalytic cycle.
In the 2014-2015 grant period, we systematically studied the potential for chain transfer between group 10 polymerization catalysts and main group elements, and we found that our initial results indicated that the rates for intramolecular chain transfer from Ni catalysts are too slow to be kinetically viable in a potential CO2/ethylene polymerization cycle. In order to increase the rates, we have focused our research efforts on synthesizing bridging, bimetallic late transition metal/main group metal complexes based on ligand frameworks that have been successful for olefin polymerization.
First, we have synthesized various mono- and bimetallic Ni complexes based on β-oxo-d-diiminate ligand frameworks (Figure 2). The monometallic complexes have a second binding pocket, of which the free 'arm' can exist as either an enamine (BODEI, β-oxo-d-enamineiminato) or imine (BODII, β-oxo-d-imineiminato) tautomer. The identity of the tautomer in the secondary Ni coordination sphere has a significant effect on ethylene polymerization behavior: the enamine tautomer, which hydrogen bonds to the central O atom and is in conjugation with the N,O backbone chelate, is significantly more electron rich and yields much lower molecular weight polymer than the imine tautomer, which rotates away from Ni to a distal position and has little effect on polymerization. Deprotonation of the second binding pocket with M(HMDS) (M = Li, Na, K) yields the Ni-alkali metal heterobimetallic complexes. The deprotonated alkali metal enamides display ethylene polymerization behavior similar to the neutral imine complex because the enamide arm can also distally rotate to minimize interaction with the Ni coordination sphere upon activation.
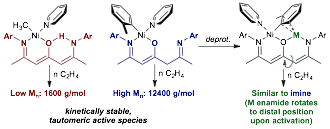
Figure 2. Ni complexes supported by β-oxo-d-diiminate ligands yield polymer that is affected by the nature of the secondary coordination sphere.
Second, we have also synthesized related complexes based on a (salicylaldimino)bipyridine framework that has a bipyridine pendant that could potentially bind a secondary metal and influence polymerization behavior (Figure 3). While ZnCl2 acts solely as a pyridine scavenger for simple imine catalyst frameworks such as biphenylimine, in the case of complexes containing a 2,2'-bipyridine pendent group, ZnCl2 can coordinate and generate a bimetallic Ni/Zn active species that produces significantly higher Mn polymer. The bipy-substituted ligand is not catalytically active in the absence of ZnCl2, and control experiments indicate that Zn coordination of the bpy pocket to generate a heterobimetallic Ni/Zn complex is critical for productive catalysis to occur. A heterobimetallic Ni/ZnCl2 precatalyst has also been synthesized and structurally characterized, and shows similar activity to the in situ bimetallic generated from ZnCl2.

Figure 3. ZnCl2 has a profound effect on ethylene polymerization with Ni salicylaldimino(bipyridine) catalysts.
Unfortunately, none of our complexes are competent for polar comonomer polymerization, and as a result we have turned back to the drawing board to explore secondary sphere effects in a more fundamental sense. Two new projects have evolved: one involving bimetallic catalyst structures for CO2 reactivity, and one involving Ir-ligand multiple bonds for C-X bond functionalization.
To this end, we have synthesized a full series of group 4/nickel complexes supported by a 2-(diphenylphosphino)pyrrolide (NP) ligand (Figure 4). Although X-ray crystallographic analysis reveals similarly short metal-metal distances in all three complexes, Quantum chemical calculations indicate that ZrNi and HfNi contain only single NiˆM dative bonds while TiNi has an additional Ti-Ni ¹-bond. We have found that the Ti-Ni bonding can also be disrupted by coordination of CO, wherein NiˆCO backbonding effectively outcompetes NiˆTi dative bonding, and we hope to use this electron reservoir in the future to cleave strong bonds such as those found in CO2.
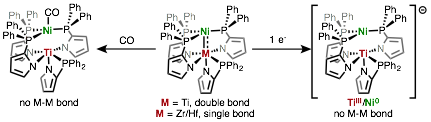
Figure 4. New heterobimetallics for CO2 activation.
Finally, we have synthesized the first terminal group 9 hydrazido(2-) complex, Cp*IrN(TMP) (TMP = 2,2,6,6-tetramethylpiperidine) (Figure 4). Electronic structure and X-ray diffraction analysis indicate that this complex contains an Ir-N triple bond, similar to Bergman's seminal Cp*Ir(NtBu) imido complex. However in sharp contrast to Bergman's imido, our complex displays remarkable redox noninnocent reactivity owing to the presence of the Nβ lone pair. Treatment of the hydrazide with MeI results in electron transfer from Nβ to Ir prior to oxidative addition of MeI to the iridium center. This behavior opens the possibility of carrying out facile oxidative reactions at a formally IrIII metal center via a hydrazido(2-)/isodiazene valence tautomerization.
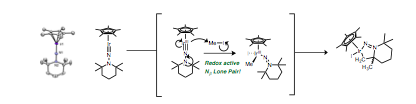
Figure 5. New redox noninnocent ligands on Ir.
Impact on Career and Students. The research performed under the ACS PRF grant window has had a profound effect on my career and my students' careers. Five students (Dr. Ryan Hue and Mr. Jimmy Chiu Mr. Peter Dunn, Mr. Adam Pearce, and Ms. Abigail Smith) have presented this research at the ACS National Conferences in Denver and Philadelphia, giving them firsthand experience presenting research projects to a broad audience. As a direct result of their research and presentations, we have set up new collaborations with Bridgestone and Exxon. The early results that have been obtained under PRF funding have also provided the initial directions for four thesis projects for students in the research group (Mr. Jimmy Chiu, Mr. Peter Dunn, Mr. Adam Pearce, and Ms. Abigail Smith).










