Reports: DNI554216-DNI5: Ethene-to-Propene Metathesis on Nickel-Exchanged Zeolites: A Fundamental Kinetic Study of the Effects of Pore Size and Nickel Structure on Ethene Dimerization Rates
 printer friendly
printer friendlyIntroduction. Steam crackers in the U.S. are shifting from petroleum-derived naphtha to shale gas-derived ethane as feedstock, which will generate excess ethene and a deficit of heavier alkenes (propene, butene) used as chemical precursors. Ethene dimerization is an entry step to upgrading of ethene to larger alkenes. The goals of this project are to study the kinetic details of ethene dimerization on Ni-exchanged molecular sieve catalysts, specifically to determine the effects of Ni active site structures, of aluminosilicate or zincosilicate supports (Fig. 1), and of the confining micropore geometry on rate constants for ethene dimerization.

Figure 1. The exchange of one extraframework Ni2+ site requires two framework Al3+ centers in a paired configuration (left; aluminosilicate) or one framework Zn2+ center (right; zincosilicate).
Current Progress. In year 2, we synthesized aluminosilicate and zincosilicate materials of the MFI, BEA, and MCM-41 framework topologies (Table 1). These materials were exchanged partially with Ni2+ cations that act as dimerization active sites, and contained either H+ (which catalyze alkene side reactions) or Li+ (which are catalytically inert) as the charge-balancing co-cations. Table 1 also summarizes representative kinetic data for ethene dimerization. Initial dimerization rates within each series of Ni-aluminosilicates (or Ni-zincosilicates) increase with pore diameter (Table 1); ongoing work to measure kinetic data on all materials in Table 1 will further clarify the effects of pore size on ethene dimerization catalysis.
Table 1. Ni-zeolites synthesized, and initial ethene dimerization rates (453 K, 0.2 kPa C2H4).
Catalyst | Pore Diameter (nm) | Si/M Ratioa | Ni/M Ratioa | Ni loading (wt. %) | Rate of C2H4 Dimerization (mol (mol Ni)-1 s-1) |
Ni-H-[Al]BEA | 0.70 | 11.0 | 0.19 | 1.20 | 1.1 x 10-3 |
Ni-Li-[Al]MFI | 0.55 | 11.8 | 0.09 | 0.60 | 2.2 x 10-4 |
Ni-Li-[Al]MCM-41 | 2.6 | 11.6 | 0.29 | 2.25 | n.m.* |
Ni-Li-[Al]BEA | 0.70 | 11.0 | 0.27 | 1.60 | 4.8 x 10-4 |
Ni-Li-[Zn]BEA | 0.70 | 4.5 | 0.16 | 1.80 | 3.5 x 10-6 |
Ni-Li-[Zn]MFI | 0.55 | n.m.* | n.m.* | n.m.* | n.m.* |
Ni-Li-[Zn]MCM-41 | 2.6 | 22.0 | 0.20 | 0.75 | 9.1 x 10-6 |
aM denotes framework metal center (Al3+ or Zn2+)
*n.m.; not measured.
On all Ni-zeolites studied, ethene dimerization rates increased with time-on-stream (activation period) to a maximum value, and then decreased with further time-on-stream (deactivation period). This transient activation period reflects structural changes to isolated Ni2+ cations to form the dimerization active site, the structural details of which are currently under investigation. Only 1-butene, cis-2-butene and trans-2-butene were formed on catalysts containing only Ni sites, while isobutene and C3 and C5+ hydrocarbons were formed on catalysts also containing residual H+ sites. Thus, double bond isomerization of butenes can occur on Ni2+ sites alone. This is an important mechanistic detail that is imprecisely understood in the experimental literature for ethene dimerization on Ni-exchanged oxides, with the majority of proposals suggesting that Ni sites form 1-butene and that residual H+ sites catalyze double bond isomerization.
This transient catalytic behavior (activation period) and the product distributions observed on Ni-zeolites are consistent with ethene dimerization via the Cossee-Arlman pathway (Figure 2), as studied recently by Brogaard and Olsbye (ACS Catal. 6 (2016) 1205) on Ni2+ cations in [Al]-SSZ-24 using density functional theory, in which the proposed active site is an ethyl coordinated Ni2+-H site and double bond isomerization steps occur on Ni-intermediates.
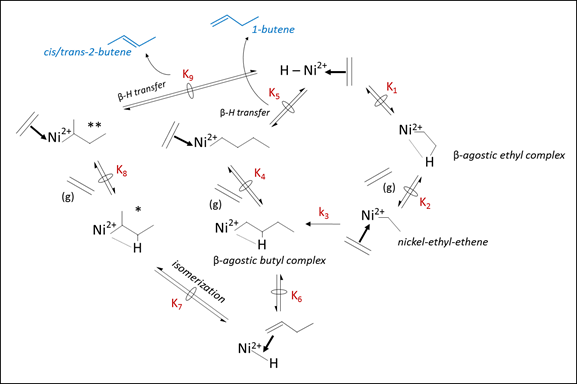
Figure 2. Cossee-Arlman pathway for ethene dimerization with proposed mechanistic assumptions on elementary step rates.
Initial ethene dimerization rates (Table 1) were estimated first by extrapolation to zero time-on-stream within a given experiment, and then by extrapolation to zero residence time from experiments performed at different space velocity. Ethene dimerization rates measured at 453 K and varying C2H4 pressures (0.01-1.0 kPa C2H4) are shown in Figure 3 for Ni-Li-[Al]BEA and Ni-Li-[Zn]BEA, and are first-order in the limit of low C2H4 pressure.
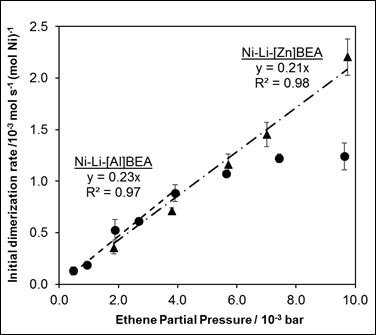
Figure 3. Initial ethene dimerization rate (453 K) dependence on ethene pressure for Ni-Li-[Al]BEA (circles) and Ni-Li-[Zn]BEA (triangles; rates multiplied by 100x for clarity). Dashed lines represent the best fit for a first-order rate law.
Brogaard et al. report that formation of a β-agostic butyl complex from nickel-ethyl-ethene intermediates (Step 3, Fig. 2) involves the highest activation barrier, implying this is the kinetically relevant step and that other steps are quasi-equilibrated. These mechanistic assumptions enable deriving the following rate expression:
![]() (1)
(1)
Furthermore, Brogaard et al. report that the β-agostic Ni-ethyl complex is the most stable nickel intermediate. Assuming this is the most abundant surface intermediate results in a rate expression that reduces to a first-order rate law (Eq. 2), consistent with our kinetic data (Fig. 3).
![]() (2)
(2)
Apparent first-order rate constants (kapp) were 0.2 and 0.002 mol s-1 (mol Ni)-1 (bar C2H4)-1 for Ni-Li-[Al]BEA and Ni-Li-[Zn]BEA, respectively. As also reflected in the initial rate data (453 K, 0.2 kPa C2H4) in Table 1, dimerization rate constants are two orders of magnitude higher on Ni sites supported on aluminosilicates than on zincosilicates. Ongoing work aims to determine the chemical origins of this phenomenon, and whether ethene adsorption energies and/or intrinsic dimerization activation enthalpies (via Eq. (2)) are different at Ni cations supported by two framework Al sites or by one framework Zn site.
Impact on Career and Participating Students. This award has enabled the PI to start a new research direction by providing full support for one graduate student (R. Joshi), and partial support for two graduate students and two undergraduate students. Joshi received a departmental scholarship (Phillips 66 Fellowship) for partial support during year 2 of this grant. The PI presented this work at the Chicago Catalysis Club Meeting (2015). Graduate and undergraduate students have presented this work within Purdue, and will present in upcoming national conferences. This project has helped generate related ideas to design catalysts with improved selectivity for other chemistries relevant for shale gas conversion, including selective dehydrogenation of light alkanes (ethane, propane) sourced from shale gas. These results are being used as preliminary data in grant proposals for shale gas upgrading to transportation fuels, submitted to government funding agencies. Thus, the research supported by this ACS PRF DNI grant is being leveraged to secure additional funds, sustain long-term research directions in our group, and continue educating the next generation of scientists and engineers.










