Reports: DNI654668-DNI6: Fundamental Investigations of Plasmons and Electron Dynamics in Petroleum-Derived Polycyclic Aromatic Hydrocarbons
 printer friendly
printer friendlyPolycyclic aromatic hydrocarbons (PAHs) are byproducts of combustion and a subject of intense scientific interest, in part due to the fact that many PAHs are known or suspected carcinogens.1 PAHs also have interesting electronic and optical properties which make them attractive as components in novel semiconducting or plasmonic devices. Repeated computational studies have demonstrated that the optical properties of the PAHs seem almost plasmonic in nature, particularly when considering dimers or trimers of PAHs.2-5 Our group has explored the ground- and excited-state properties of these materials to assess their ability to mimic the properties of conventional plasmonic nanomaterials. We approached this problem from two directions. First, we developed and applied real-time time-dependent quantum-mechanical approaches in order to understand the ultrafast dynamics of electrons in PAHs. Second, we developed novel electronic structure methods to accurately describe the complex nature of both the ground and excited states of these materials.
We have used explicitly time-dependent electronic structure methods to explore the ultrafast dynamics of electrons in PAHs. We developed an implementation of the time-dependent (TD) configuration interaction singles (CIS) method within the Psi4 electronic structure package that allows us to assess the ability of PAHs to mimic the properties of plasmonic materials. In Figure 1, we consider the interaction of a dimer of naphthalene molecules with incident electromagnetic radiation that is resonant with the bright longitudinal excitation in naphthalene at roughly 6.7 eV (as predicted by CIS/aug-cc-pVDZ). The system undergoes Rabi flopping, and the induced time-dependent electric fields show the characteristic beating usually observed in the dipole moment during the Rabi cycle. Figure 1 (b) illustrates the enhancement in the electric field between two naphthalene molecules as a function of their separation, which approaches a factor of 20 at small intermolecular distances.
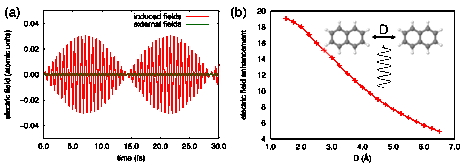
Figure 1. For naphthalene dimer under resonant excitation, the (a) induced time-dependent electric fields and (b) the enhancement in the electric field.
We also developed time-domain electronic structure methods beyond TD-CIS that could provide higher-quality descriptions of the dynamics of electrons in PAHs. We first developed an implementation of time-dependent Hartree-Fock (TDHF) theory for this purpose. We discovered that a molecule modeled with TDHF could easily be coupled to a cavity quantum electrodynamics (CQED) based description of a fully-developed plasmon resonance for a metallic nanoparticle (MPN).6 This combined CQED/TDHF formalism was used to model coupled plasmon-molecule interactions and demonstrated the emergence of a Fano-like resonance that was quite sensitive to the MNP-molecule separation. We are working to formulate a version of the theory to model the injection of hot electrons generated by the decay of the plasmon into nearby PAHs or other molecules. We also developed a time-dependent coupled-cluster code based on the equation-of-motion (EOM) approximate second-order coupled cluster (CC2) model.7 We demonstrated that linear absorption spectra for large molecules with a high density of states can be obtained from a single TD-EOM-CC2 simulation [see Fig. 2]. We are exploring the utility of TD-EOM-CC2 in the description of X-ray Absorption Near Edge Structure (XANES) for PAHs and other small molecules.
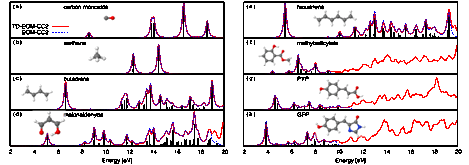
Figure 2 Normalized and artificially broadened absorption spectra computed with the TD-EOM-CC2.
While single-reference approaches like TD-CIS, TDHF, or TD-EOM-CCC2 can provide an adequate description of electron dynamics in small PAHs, larger PAHs, whose ground states have substantial multireference character, are not necessarily well described at these levels of theory. The usual workhorse for capturing multireference correlation is the complete active space self-consistent field (CASSCF) method. Unfortunately, most implementations of CASSCF are configuration-interaction (CI)-based, and the exponential scaling of CI severely limits the size of the active space that one can practically employ. The application of CASSCF to large active spaces requires that one abandon CI in favor of polynomial-scaling approaches such as density-matrix renormalization group (DMRG)8 or variational two-electron reduced-density matrix (v2RDM) methods.9 We developed an implementation of the v2RDM method10 and coupled it to an orbital optimization procedure, thereby achieving polynomial-scaling CASSCF.11 We applied v2RDM-CASSCF to the ground-state properties of the linear acene series; we assessed the ability of the v2RDM-CASSCF method to capture the emergence of polyradical behavior in longer members of this series and the ability of the method to predict the energy gaps between the lowest-energy singlet and triplet states (see Fig. 3). The v2RDM-CASSCF description of the electronic structure is similar to that provided by other sophisticated methods, such as DMRG. The largest v2RDM-CASSCF computations considered here involved an active space of 50 electrons in 50 orbitals and the simultaneous optimization of 1896 orbitals. Such a computation is impossible with CI-driven CASSCF.
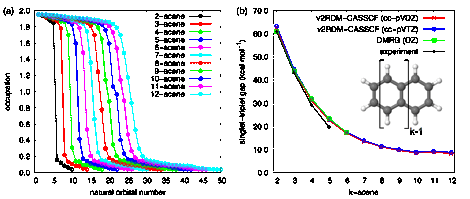
Figure 3. For the linear acene series, (a) natural orbital occupation numbers convey the emergence of polyradical behavior. (b) The v2RDM-CASSCF singlet/triplet splittings agree well with those from experiment and DMRG (taken Ref. 12).
The impact of this work on my career and that of my students has been significant. This grant has supported work that led to three published manuscripts,6,11,12 and work described in a fourth manuscript is currently under review.7 Importantly, we have generated a substantial amount of data that has served as preliminary data for other proposals to federal funding agencies. The graduate students working on this project have learned valuable research skills, particularly computer programming skills, which are transferable to virtually any other discipline.
[1] Luch, A., 'The Carcinogenic Effects of Polycyclic Aromatic Hydrocarbons.' Imperial College Press: London, 2005.
[2] Manjavacas, A.; et al., ACS Nano 2013, 7, 3635-3643.
[3] Guidez, E. B.; Aikens, C.M. J. Phys. Chem. C 2013, 117, 21466-21475.
[4] Bursi, L.; Calzolari, A.; Corni, S.; Molinari, E. ACS Photonics 2014, 1, 1049-1058.
[5] Lauchner, A.;, et al., Nano Lett. 2015, 15, 6208-6214.
[6] Nascimento, D.R.; DePrince, A.E. J. Chem. Phys. 2015, 143, 214104.
[7] Nascimento, D.R.; DePrince, A.E. under review 2016.
[8] Ghosh, D.; et al. J. Chem. Phys. 2008, 128, 144117.
[9] Gidofalvi, G.; Mazziotti, D.A.; J. Chem. Phys. 2008, 129, 134108.
[10] Fosso-Tande, J.; Nascimento, D.R.; DePrince, A. E. Mol. Phys. 2016, 114, 423-430.
[11] Fosso-Tande, J.; Nguyen, T.-S.; Gidofalvi, G.; DePrince, A.E. J. Chem. Theory Comput. 2016, 12, 2260-2271.
[12] Hachmann, J.; et al. J. Chem. Phys. 2007, 127, 134309.










