Reports: ND755833-ND7: Sterically-Driven Selectivity in Acyclic Diene Metathesis (ADMET) Polymerization of Asymmetric alpha,omega-Dienes for Sequence-Controlled Polyolefins
 printer friendly
printer friendlyADMET Monomer Synthesis
The objective of this proposal is the investigation of sterically-driven cross metathesis selectivity as an innovative approach for the synthesis of sequence-controlled, regioregular polyolefins via ADMET polymerization of asymmetric α,ω-diene monomers. Our initial synthetic efforts have focused on the preparation of asymmetric α,ω-diene monomers comprising an unhindered Type I α-olefin and a hindered Type III ω-terminal protected tertiary allylic alcohol – where the degree of steric hindrance around the ω-terminal olefin will be varied by altering the moiety used for protection of the allylic alcohol. 1,2-epoxy-7-octene (1) served as a commercially available precursor for the asymmetric α,ω-diene monomers. As shown in Figure 1, 1 was ring-opened with lithium triethylborohydride in THF to yield the intermediate 7-octen-2-ol (2) in 89% yield after purification via flash chromatography. 2 was then oxidized to the ketone 7-octen-2-one (3) using pyridinium chlorochromate (PCC) in dichloromethane (78% yield). 3 was finally converted to the tertiary allylic alcohol (4) via a Grignard reaction with vinylmagnesium bromide in THF.
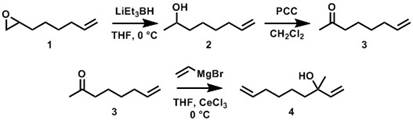
Figure 1. Synthesis of asymmetric α,ω-diene containing a Type I alkene and a Type II/III tertiary allylic alcohol.
Initially, the Grignard reaction with 3 resulted in many side products from competing reactions (e.g., enolization, conjugate addition). To overcome these undesired side products which resulted in poor yields of the desired product, we explored cerium chloride promoted Grignard reactions to suppress these side reactions. The CeCl3 promoted Grignard reaction provided 4 in 70% yield following purification via column chromatography. The structures of all compounds were confirmed by 1H (Figure 2) and 13C NMR.
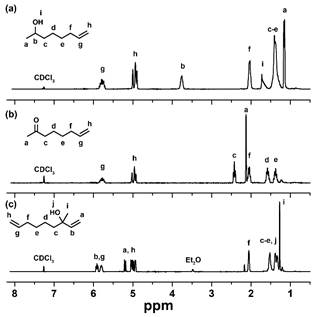
Figure 2. 1H NMR spectra of (a) 7-octen-2-ol, (b) 7-octen-2-one, and (c) tertiary allylic alcohol asymmetric diene.
Thiol-TFVE Chemistry
Support from the current PRF grant has also enabled the exploration of thiol-trifluorovinyl ether (thiol-TFVE) chemistry. Here, we report a first example of thiol-trifluorovinyl ether (thiol-TFVE) photopolymerization as a facile, cure-on-demand synthetic route to semi-fluorinated polymer networks.
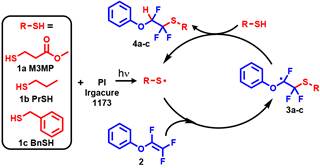
Figure 3. Model reaction between Ph-TFVE and a series of thiol monomers.
The thiol-TFVE reaction was initially investigated using model reactions between phenyl trifluorovinyl ether (Ph-TFVE) and a series of thiols of increasing reactivity (methyl 3-mercaptopropionate [M3MP] > 1-propanethiol [PrSH] > benzyl mercaptan [BnSH]), as shown in Figure 3. TFVE, thiol, and photoinitiator were combined without solvent and exposed to UV
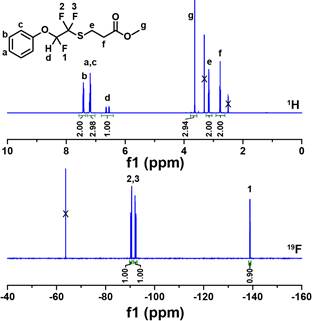
Figure 4. 1H NMR (top) and 19F NMR (bottom) of M3MP/Ph-TFVE addition adduct.
light (100 mW/cm2) in air. The structure of the thiol-TFVE adduct was characterized using 1H and 19F NMR. Figure 4 shows the 1H and 19F NMR spectra for the M3MP/Ph-TFVE adduct. In each reaction, the thiol successfully added across the TFVE functional group to afford a semi-fluorinated ether/thioether linkage. Control experiments conducted with M3MP/Ph-TFVE in the absence of photoinitiator and UV light revealed no reaction after 5 days suggesting the thiol addition does not proceed through charge transfer or co-oxidation reactions. The appearance of a single proton peak, d, centered around 6.50 ppm represents the hydrogen on the carbon alpha to the ether linkage, previously a part of the thiol functional group. Additionally, 19F NMR reveals the disappearance of the TFVE group and the formation of two doublets of doublets of doublets (ddd) and a doublet of triplets (dt), representing fluorines 1 and 2, and 3, respectively. The splitting patterns and associated integration values were used to confirm the anti-Markovnikov addition of thiols to the TFVE.
Thiol-TFVE Photopolymerization for Semi-Fluorinated Networks
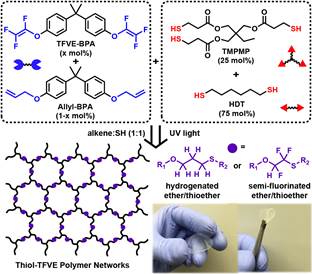
Figure 5. Thiol/TFVE multifunctional network monomers and network preparation.
The rapid and efficient reaction kinetics between the TFVE and a variety of thiols make the thiol-TFVE photoreaction an ideal candidate to rapidly synthesize semi-fluorinated polymer networks with highly tunable properties. Therefore, a difunctional TFVE monomer based on bisphenol A (TFVE-BPA) was synthesized and formulated with di- and trifunctional thiol monomers, as shown Figure 5. TFVE-BPA was copolymerized with allyl-BPA to tailor the amount of semi-fluorinated linkages in the network and determine the effect of the TFVE group on the photopolymerization process. The structural similarity of allyl-BPA serves as a control to determine the influence of the semi-fluorinated linkage on polymer network properties. All formulations contained 1 wt.% Irgacure 1173 and were polymerized with 20 mW/cm2 UV light under an inert N2 atmosphere. The mol % of TFVE groups was systematically varied from 0 mol % to 100 mol %. The polymerization kinetics as a function of TFVE-BPA concentration are shown in Figure 6. After 2 minutes of UV exposure, all network formulations reached near quantitative conversion with stoichiometric conversion of all functional groups. These results confirm that under inert atmosphere, semi-fluorinated networks can be rapidly synthesized, and that the TFVE group exerts little to no influence on final conversion values.
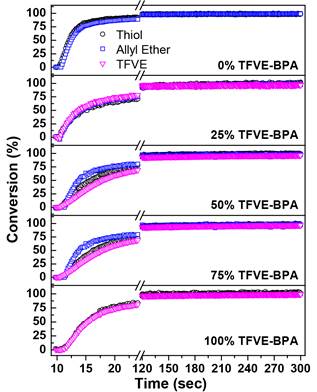
Figure 6. FTIR conversion plots of thiol-TFVE networks formulated with 0-100 mol % TFVE-BPA.
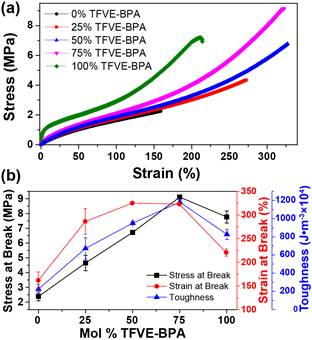
Figure 7. (a) Stress/strain curves of 0-100 mol % TFVE-BPA thiol/TFVE networks (b) Summation of thiol/TFVE polymer network tensile properties.
The influence of the semi-fluorinated ether/thioether linkage is also evident in the mechanical properties of the thiol-TFVE networks. Representative stress-strain curves are shown in Figure 7a and summarized in Figure 7b. In general, both stress at break and strain at break values increased with increasing TFVE-BPA concentration, with the 50-75% TFVE-BPA networks exhibiting a 2-fold increase in strain at break relative to the 0% TFVE-BPA sample. It is also clear that increasing the concentration of semi-fluorinated linkage within the network results in a 5-fold increase in toughness, peaking at 75 mol % TFVE, relative to the non-fluorinated network (0% TFVE). The 100% TFVE network showed a significant increase in initial modulus; however, a decrease in toughness was observed relative to the 50-75% TFVE samples. The decrease is toughness can be attributed to the more glass-like behavior of the 100% TFVE, as tensile testing was conducted at a temperature (25 °C) just below Tg. The semi-fluorinated linkage provides hydrogen bonding sites as physical crosslinks that are capable of absorbing the energy of mechanical deformation and rupturing before the covalent crosslinks. Ultimately, incorporating semi-fluorinated ether-thioether linkages via thiol-TFVE photopolymerization provides a facile route to enhance the mechanical properties of thiol-ene polymer networks.










