Reports: ND655476-ND6: Metal-H2 Complexes vs Metal Hydrides: Nuclear motion and implications for hydrogenation catalysis
 printer friendly
printer friendlyIntroduction
The objective of this research program is to investigate the role of molecular motion in metal-bound H2 complexes and metal hydrides. These species, which play a central role in catalytic hydrogenation processes, serve as exquisite examples of the coupling between unique electronic structure and dynamical nuclear motion. The research program seeks to unravel the details of this coupling, as well as the fundamental chemical implications of the resulting motion, through modern, quantum mechanics-based computer simulations.
Significance
The long-term question to be addressed by this new research program (and beyond) is as follows: What is the optimal level of dihydrogen/hydride interconversion for hydrogenation catalysts? Motivating this question is the observation that metal-dihydrogen complexes possess a reasonably intact H-H bond with relatively weaker (15-20 kcal/mol) bonding of the dihydrogen moiety to the metal center. By contrast, canonical hydride complexes exhibit an effectively broken H-H bond, but the H-H bond is stronger (30-40 kcal/mol). Does the latter class of compounds undergo movement within the coordination sphere more easily? Or does the ligand-exchange propensity of complexes intermediate to these regimes help with intra-sphere mobility?
This PRF-funded research program is investigating the nature of bonding and molecular structure—or lack thereof—in metal-dihydrogen/hydride complexes. Historically, focus has rightfully been placed on electronic contributions to the bonding, but the resulting molecular motion is a key contributor to structure, thermodynamics, and reaction mechanisms. Importantly, such studies can now be performed without ignoring the important role of accurate electronic structure theory. The main significance of the work in this grant period is a new, complete picture of fluxional structure in these complexes, as well as a conceptual explanation of the electronic sources of this motion.
Year 1 Progress
The research program to-date has dominantly focused on three representative complexes, as well as technical improvements in the underlying computations in order to make these simulations tractable. As a simple but illustrative example of catalytic behavior in the presence of both sigma‑H2 and hydride forms, the PdH3- molecule was first studied. The much larger complex, Mo(PH3)5H2, was also studied as a model of more realistic catalysts and a system in which H2 and cis-/trans-dihydride forms are all accessible. Finally, an undergraduate has performed preliminary calculations on FeH(H2)(PMe3)4+, which will represent the much more fluxional regime of mixed hydride/sigma complexes. Each of these complexes and current results will be discussed, in turn, below.
1. PdH3-
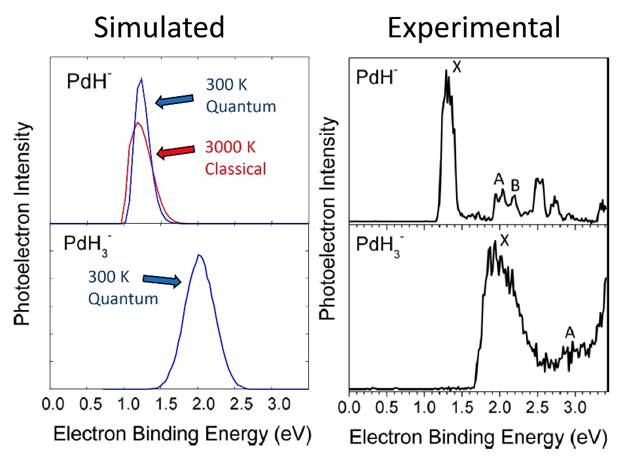
Photoelectron spectra of the mixed sigma/hydride complex PdH3- were recently reported by Bowen and coworkers. Given that this complex could potentially exhibit exactly the H2/hydride interchange dynamics that we sought to examine in this research program, we elected to examine its quantum structure (electronic and vibrational) in more detail. The discussion in the original experimental paper was reasonably based on electronic structure at equilibrium. However, after further investigation, we discovered a low barrier to interconversion and scrambling of the hydrogenic moieties. A bare H2 molecule requires approximately 100 kcal/mol to break its chemical bond, yet rupture of this same bond at the hydrogen-transfer transition state of PdH3- was found to be only 6 kcal/mol uphill from equilibrium.
Our analysis of this complex was two-fold:
(a) Electronic structure – The main property addressed in this analysis is the electronic source of the stability of the hydrogen/hydride-transfer transition-state structure. After careful benchmarking with high-level coupled-cluster calculations, the BP86 density functional was selected as a reliable compromise of accuracy and efficiency. Using this method, the equilibrium complex exhibited hallmark orbital interactions, including dz2-sigma forward-donation and dxz-sigma* back-donation effects. More interestingly, as the H was passed around the metal center during the H/H2-to-H2/H migration, other d orbitals were able to appreciably stabilize the moving hydrogen.
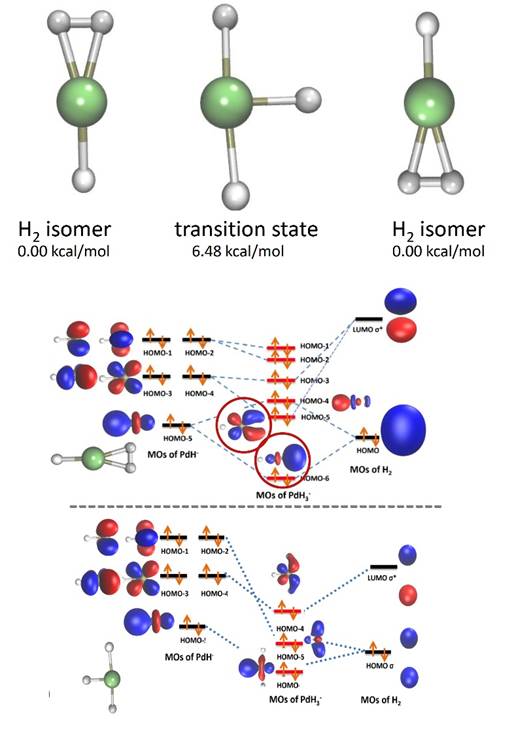
(b) Molecular motion – Using on-the-fly ab initio path integral molecular dynamics (PIMD) techniques, the quantum motion of this complex was investigated. As a secondary calibration of our electronic structure method, the photoelectron spectra were computed from these simulations. Through vertical ionization of the PIMD distribution, good consistency with the experimental breadth of the photoelectron signal was obtained; this breadth was found to be dominantly due to inherent molecular motion. The hydrogen-exchange mechanism exhibited a high enough barrier that PIMD simulations were not straightforward, however. Instead, we opted to fit an analytic potential energy surface, using permutationally invariant polynomials. After careful fitting, we were able to access much longer timescales and directly observed the barrier-crossing dynamics. Because of the height of the barrier, the dynamics are dominantly over-barrier in nature, rather than quantum tunneling, although the latter does contribute at lower temperatures. Remaining tasks include the development of a more accurate potential surface and final simulation of this complex at multiple temperatures.
2. Mo(PH3)5H2
This second-row transition-metal complex was selected due to previous computational studies that suggested low energy penalties to H2/hydride isomerization. In fact, using much more accurate quantum chemistry methods, we found that the structures were even closer in energy and may be considered effectively degenerate. More interestingly, when harmonic zero-point energy corrections were included, the barrier between the sigma-H2 and cis-dihydride form became negative, suggesting the possibility of particularly unique quantum motion, and these types of motions are of considerable interest in this study.
To simulate a complex of this size in full dimensionality, with quantum chemistry potentials and including nuclear quantum motion effects, improvements in our existing computational framework were required. Technical improvements included the implementation of a mixed OpenMP-OpenMPI parallelization for the PIMD simulations. Furthermore, access to barrier heights (including assessment of quantum effects) required the implementation of enhanced-sampling techniques during the PIMD simulations. Such improvements have been implemented and are now being used to simulate this much larger complex.

3. FeH(H2)(PMe3)4+
An undergraduate student has performed preliminary quantum benchmarking calculations on the –PH3 analogue of this iron(IV)-based complex. This system is of special interest, due to the potential mixed sigma/hydride dynamics (based on early studies), as well as the role of neighboring ligands in enhancing or restricting this quantum motion, compared to the less hindered PdH3- complex, discussed above. Full-dimensional simulations of this complex will be performed this fall.











