Reports: DNI655229-DNI6: Study of the Support to Active Site Interaction in Catalysts of Oil Hydrocracking and Hydrodesulfurization Using New Density Functional Theory Methods
 printer friendly
printer friendlyImprovement of applicability of new computational methods
The catalytic processes such as hydrocracking (HC) and hydrodesulfurization (HDS) involve transition-metal centers such as MoS2 and Co-MoS2 supported on g-Al2O3 and TiO2surfaces. These are large molecular structures involving many atoms including heavy ones. They are particularly challenging computationally for new DFT methods because the latter have higher computational cost than conventional DFT methods. The reason is that the new methods require the computation of the HF (Hartree-Fock) exchange energy density. Its computation is expensive and complicates the derivation of SCF solution. We originally proposed a general solution based on RI (resolution of identity) previously for this problem. Our RI solution allowed us to achieve the SCF solution for B05 for the first time, but it is still inefficient especially for large systems, and numerically unstable.
Instead of approximating, we have recently developed an algorithm to compute the original integrals directly and exactly. The details will be published elsewhere but the core idea is that the electrostatic potential (ESP) for each basis function pair, is calculated first at each grid point. This scheme scales the same as the normal HF exchange energy with respect to the molecular size and thus scales better than RI for large molecules. Furthermore, it requires much less memory than the original RI scheme.
We compared the performance of the new scheme with that of RI for an SCF calculation with B05 functional for a number of molecules of different sizes with the popular 6-31G** basis set. The timings are for one SCF iteration in seconds. As one can see, the new scheme is faster than RI, and the gain of performance grows as the molecular size increases. The new scheme is also numerically more stable.
Method |
Pyrrole |
Benzene |
Alanine |
Naphthalene |
New scheme |
42 |
67 |
79 |
164 |
RI scheme |
57 |
98 |
121 |
400 |
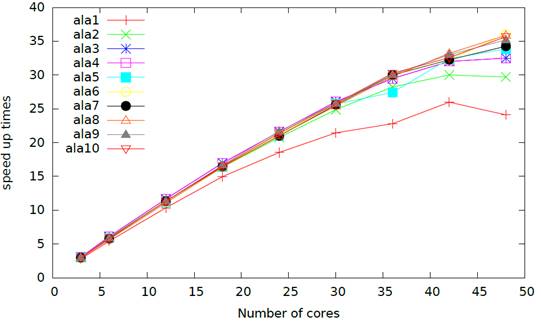
To further improve the applicability of our methods, we implemented our code on multicore computers. The figure below shows the parallel efficiency of our implementation on a series of alanine peptides. As one can see, our code scales very well with respect the number of cores, achieving almost linear speed-up for the larger alanines.
Studies on the ethlylene addition to Ni dithiolene reactions
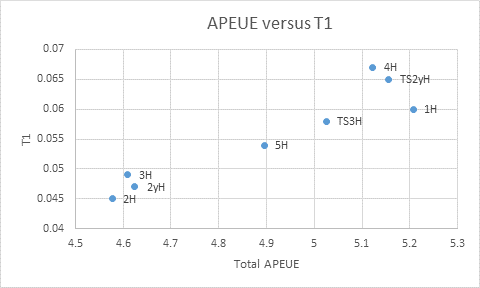
To assess the applicability of our methods for catalytic reactions, we tested our method for the computation of atomic population of effectively unpaired electrons (APEUE) on the ethylene addition to a Ni-S center Ni(edt)2 (edt=S2C2H2) [4]. We computed the APEUE numbers on various reactants, products and transition states as listed in the reference [4], and compared the numbers of T1 analysis, an index based on a wavefunction method CCSD(T). CCSD(T) is a high-level method and its results are considered reliable. The graph below shows the correlation between the indexes computed with the two methods. As one can see, they correlate rather well. The advantage of our method is that it is computationally much more affordable than CCSD(T) since it is a DFT-based method. This gives us confidence at applying our methods to studying catalysis.
References
1. Becke, A.D., Real-space post-Hartree-Fock correlation models. Journal of Chemical Physics, 2005. 122: p. 64101.
2. Liu, F., et al., Comparison of the performance of exact-exchange-based density functional methods. Journal of Chemical Physics, 2012. 137: p. 114104.
3. Proynov, E., F. Liu, and J. Kong, Analysing effects of strong electron correlation within Kohn-Sham Density-Functional Theory. Phys. Rev. A, 2013. 88: p. 032510.
4. Dang, L., et al., Computational studies on ethylene addition to nickel bis(dithiolene). J. Phys. Chem. A., 2012. 116.










