Reports: UNI354833-UNI3: Multifunctional Bipyridine Ligands for the Catalytic Reduction of CO2 to CO
 printer friendly
printer friendlyCarbon monoxide (CO) can be used as a starting material for many valuable chemicals. One promising way to produce CO is via the reduction of carbon dioxide (CO2) produced from petroleum usage. However, reduction of CO2 to CO is a formidable challenge, and a suitable catalyst is required for this reaction to occur with enough speed, efficiency and selectivity. When [M(CO)3(R-bpy)X] complexes (M = Re or Mn, R-bpy = bipyridine or bipyridine derivative, and X = halide) are reduced in situ they can catalyze reduction CO2 to CO with good selectivity.1 For most CO2 reduction catalysts of this type, catalysis occurs at or near the second of two one-electron reduction events. The reduction pathway involves protonation of CO2, and recently addition of an intramolecular Brønsted acid in the form of a phenol substituent on the bipyridine ligand of a [Mn(CO)3(R-bpy)Br] catalyst was found to result in an enhanced catalytic current at a moderate overpotential.1 Support from the PRF is enabling us to further investigate the effect of pKa and positioning of this intramolecular Brønsted acid substituent on the rate and efficiency of CO2 reduction catalysis. This research involved numerous undergraduate students at the University of Wisconsin Oshkosh, all of whom experienced fundamental scientific research for the first time through working on this project.
The first line of inquiry consisted of synthesis of the complexes [Mn(CO)3(6,6’-dihydroxy-2,2’-bipyridine)Br] and [Re(CO)3(6,6’-dihydroxy-2,2’-bipyridine)Cl]. Both compounds were characterized spectroscopically and crystallographically (Figure 1), and were determined to be pure by elemental analysis. CO2 coordinates to the metal center at a site cis to the bipyridyl ligand, so the hydroxyl groups are not positioned to interact directly with the metal-bound CO2 substrate, but they could potentially facilitate protonation of the bound CO2 by creating a local source of protons and through hydrogen-bonding with water present in the reaction solution. Cyclic voltammetry (CV) was performed on [Mn(CO)3(6,6’-dihydroxy-2,2’-bipyridine)Br] and, for comparison, [Mn(CO)3(bipyridine)Br] under an inert atmosphere (argon) and under catalytic conditions (CO2 atmosphere and 5% water) (Figure 2). [Mn(CO)3(6,6’-dihydroxy-2,2’-bipyridine)Br] lacks reversible or quasi-reversible electrochemical processes, complicating a determination of its turnover frequency under catalytic conditions, but the onset of the catalytic wave for [Mn(CO)3(6,6’-dihydroxy-2,2’-bipyridine)Br] (-2.36V vs. ferrocene/ferrocenium (Fc/Fc+)) is significantly more negative than that of [Mn(CO)3(bipyridine)Br] (-1.99V vs. Fc/Fc+), indicating that the former has a significantly higher overpotential and lower efficiency. Controlled potential electrolysis coupled with gas chromatography must be performed to determine the species produced from this catalytic current.
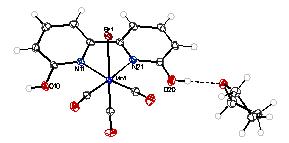

Figure 1. Crystal structures of [Mn(CO)3(6,6’-dihydroxy-2,2’-bipyridine)Br] (left) and [Re(CO)3(6,6’-dihydroxy-2,2’-bipyridine)Cl]
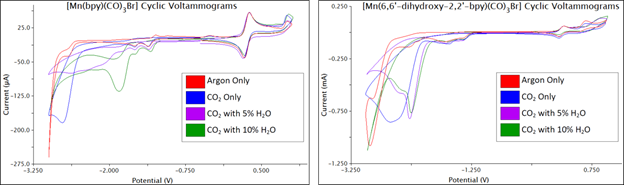
Figure 2. CVs performed in acetonitrile (MeCN) solvent, 0.1M tetrabutylammonium PF6 (TBAPF6) electrolyte, and referenced to ferrocene/ferrocenium
A second line of inquiry is an investigation of the effect of the pKa of the intramolecular Brønsted acid on CO2 reduction rate, overpotential, and selectivity. We began by synthesizing a ligand in which the phenol substituent studied previously2 was replaced by a benzoic acid substituent. The pKa of phenol and benzoic acid in acetonitrile have been determined to be 29.14 and 21.51,3 respectively, so the ligand containing the benzoic acid substituent would be expected to be significantly more acidic than the ligand containing the phenol substituent. The synthetic route for the benzoic acid-containing ligand, termed 6-PhCOOH-2,2’-bpy, is shown in Scheme 1.
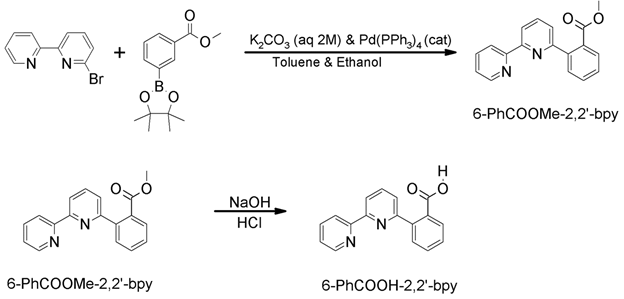
Scheme 1. Synthesis of 6-PhCOOH-2,2’-bpy ligand.
Metallation with MnBr(CO)5 and ReCl(CO)5 yielded the complexes [Mn(6-PhCOOH-2,2’-bpy)(CO)3Br] and [Re(6-PhCOOH-2,2’-bpy)(CO)3Cl], respectively. Thus far, these complexes have been characterized spectroscopically, crystallographically (Figure 3), and by CV. Interestingly, whereas CV’s of similar complexes exhibit catalytic waves at or near the second reduction peak in the presence of CO2 and water, for these complexes no current enhancement is observed until after a third reduction event, at much more negative potentials than is typically observed (Figure 4). Moreover, this current enhancement has been found to be scan rate-dependent up to scan rates of 1.5 V/s. Further experimentation will be done to elucidate why catalysis is not observed following the second reduction event and to determine the energetics for rotation of the benzoic acid substituent about the metal center.

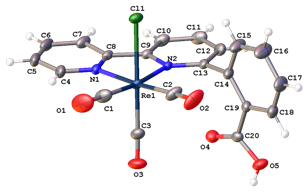
Figure 3. Molecular drawings of [Mn(6-PhCOOH-2,2’-bpy)(CO)3Br] (left), with the minor disorder component shown with dashed lines), and [Re(6-PhCOOH-2,2’-bpy)(CO)3Cl] (right). Both complexes are drawn with 50% probability ellipsoids and have the solvent and hydrogen atoms omitted. Data collections and analyses were performed by Ilia Guzei at University of Wisconsin Madison.
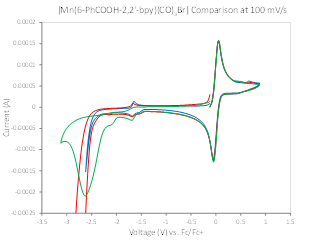
Figure 4. CVs performed in acetonitrile (MeCN) solvent, 0.2M tetrabutylammonium PF6 (TBAPF6) electrolyte, and referenced to ferrocene/ferrocenium, under a N2 atmosphere (blue trace), under N2 with 5% water (red trace), and under CO2 with 5% water (green trace).
Since the benzoic acid substituent resulted in inhibition of catalysis at potentials where it is normally observed, we synthesized a different ligand to enable us to study the effect of pKa of the intramolecular acid substituent on catalysis, in which fluorine is added to a phenol substituent to lower the pKa of the phenol substituent. This ligand is termed F-HOPh-bpy, and is shown in Figure 5 along with the previously utilized ligand HOPh-bpy.2 F-HOPh-bpy was synthesized via a procedure similar to that for 6-PhCOOH-2,2’-bpy. Metallation with MnBr(CO)5 and ReCl(CO)5 yielded the complexes [Mn(F-HOPh-bpy)(CO)3Br] and [Re(F-HOPh-bpy)(CO)3Cl], respectively, which have been characterized spectroscopically. Preliminary CV experiments indicate that [Mn(F-HOPh-bpy)(CO)3Br] exhibits a catalytic current that is moderately higher than the previously reported catalyst [Mn(F-HOPh-bpy)(CO)3Br] at a slightly more positive potential. In the future, we will complete the characterizations of both [Mn(F-HOPh-bpy)(CO)3Br] and [Re(F-HOPh-bpy)(CO)3Cl], including complete electrochemical characterizations in order to determine the catalytic rates, overpotential, and selectivity. We will compare their catalytic performances to those of [Mn(HOPh-bpy)(CO)3Br] and [Re(HOPh-bpy)(CO)3Cl] in order to assess the effect of the pKa of an intramolecular acidic substituent on catalytic performance, which will provide valuable information regarding catalysis design and maximizing catalytic performance.

Figure 5. F-HOPh-bpy (left) and HOPh-bpy (right)
1. (a) Takeda, H. et al. JACS 2008, 130 (6), 2023–2031; (b) Bourrez, M. et al. Angewandte Chemie Int. Ed. 2011, 50 (42), 9903-9906.
2. Agarwal, J. et al. Inorg. Chem. 2015, 54, 5285-5294.
3. Muckerman, J.T. et al. Biochim. Biophys. Acta 2013, 1827(8-9), 882-91.










