Reports: UNI153983-UNI1: Synthesis and Cycloadditions of Alkyl-substituted Siloles and Exploration of Strained Bicyclic Silane Reactivity
 printer friendly
printer friendlyDuring the final year of support from the ACS Petroleum Research Fund, we began by following up on our observation that bridged tricyclic silanes (1) derived from 2,5-diphenyl-substituted siloles were hydrolytically unstable, opening to silanols during column chromatography (Scheme 1). This is in contrast to bridged tricyclic silanes (2 and 3) made from 3,4-dialkyl-substitued siloles, which may be isolated via chromatography and stored indefinitely.1
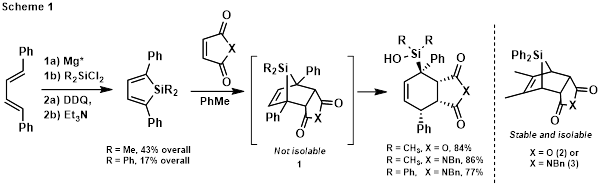
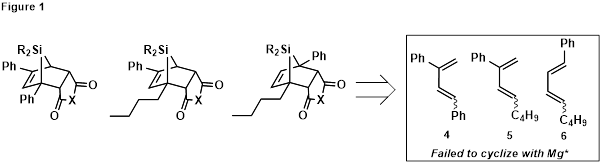
We initially sought to synthesize bridged polycyclic silanes with different substitution patterns, having both alkyl and aryl substituents, to compare their stabilities and reactivity (Figure 1). However, dienes 4-6 failed to cyclize with Rieke Mg (Mg*),2 blocking access to the requisite siloles by this route.
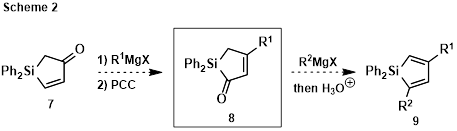
A new approach to substituted siloles was thus sought, enabling flexible late-stage introduction of substituents. Compound 8 (Scheme 2) was viewed as a promising intermediate that might be derived from enone 7 via Grignard addition, followed by oxidative rearrangement with PCC. Intermediate 8 could then converted to disubstituted siloles 9.
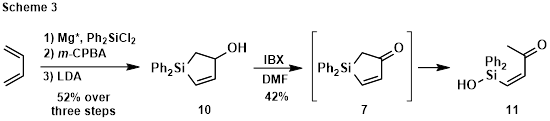
Synthesis of enone 7 was pursued as shown in Scheme 3. Allylic alcohol 10 was made via Pagenkopf’s procedure,1 and then oxidized using IBX. Unfortunately, ring-opened silanol 11 was isolated, resulting from hydrolysis of the desired enone. Other oxidants also failed to deliver the desired product.
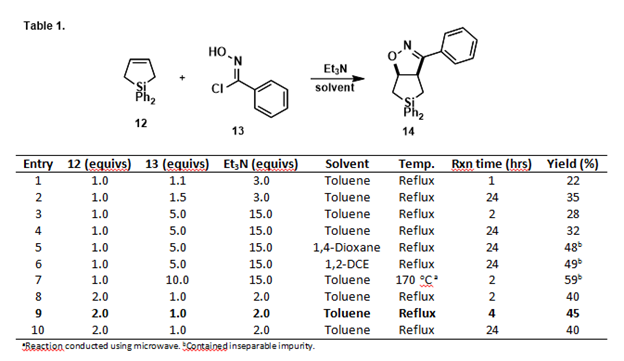
The susceptibility of cyclic silanes to C-Si bond cleavage introduces significant challenges to their use in multi-step synthesis. Indeed, both our efforts to explore novel applications of bridged polycyclic silanes and our efforts to synthesize silacyclopentenones for conversion to siloles were thwarted by undesired ring-opening hydrolysis of C-Si bonds. However, many silicon-containing compounds have desirable medicinal properties,3 prompting us to focus our studies on an exploration of [3+2] cycloadditions of dihydrosilole 12 with nitrile oxides to form isoxazoline-fused silacyclopentane bicycles 14 (Table 1). These compounds were viewed as versatile intermediates that might be transformed to a variety of useful structures if appropriately chemoselective reductions were developed. Our goal was, therefore, to study the susceptibility of the C-Si bonds to cleavage while attempting to chemoselectively reduce the N-O and/or C=N bonds.
Cycloaddition of 12 with the nitrile oxide derived from imidoyl chloride 13 was investigated under a variety of conditions as shown in Table 1. The best combination of yield and purity was obtained using 12 in excess, refluxing in toluene for 4 hours (Entry 9). Employing other imidoyl chlorides under the same conditions afforded the family of fused bicyclic silanes shown in Figure 2.
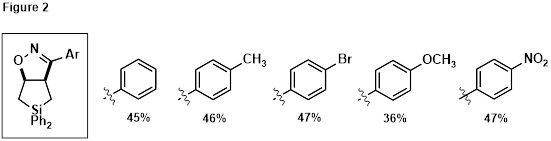
To our initial surprise, subjecting fused bicycles 14 to LiAlH4 afforded oximes 15 (Scheme 4, configuration unconfirmed), resulting from hydride attack at silicon, C-Si bond cleavage, and elimination to open the isoxazoline ring. Good-to-excellent yields were obtained, leaving the C=N and N-O bonds intact. This result is consistent with the reactivity observed during the oxidation of allylic alcohol 10 (Scheme 3), confirming that C-Si bond cleavage is facile when an electron accepting/leaving group is present at C3 of the silacyclopentane.
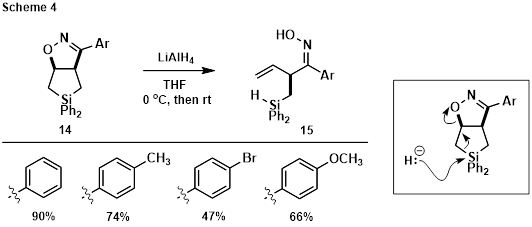
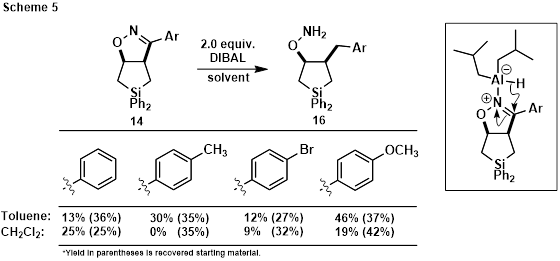
Reasoning that DIBAL would coordinate to one of the isoxazoline heteroatoms prior to hydride transfer, reduction of the C=N bond without C-Si bond cleavage was also attempted (Scheme 5). Double hydride addition to the C=N bond was observed under these conditions, delivering hydroxylamine derivative 16 in which the C-N bond was cleaved, leaving the N-O and C-Si bonds intact. Yields were modest indicating further optimization is required.
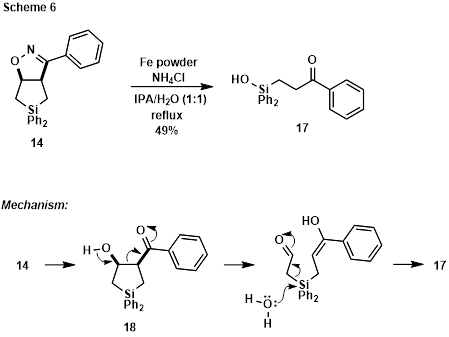
The final set of reduction conditions briefly explored in this survey employed Fe powder and NH4Cl.4 Refluxing 14 in IPA/H2O under these conditions afforded b-keto silanol 17. Mechanistically, N-O bond cleavage, followed by hydrolysis of the resulting imine would deliver b-hydroxy ketone 18. Retro-aldol reaction, followed by water-mediated C-Si bond cleavage, would facilitate extrusion of acetaldehyde in enol form to deliver 17 (compare to Scheme 3).
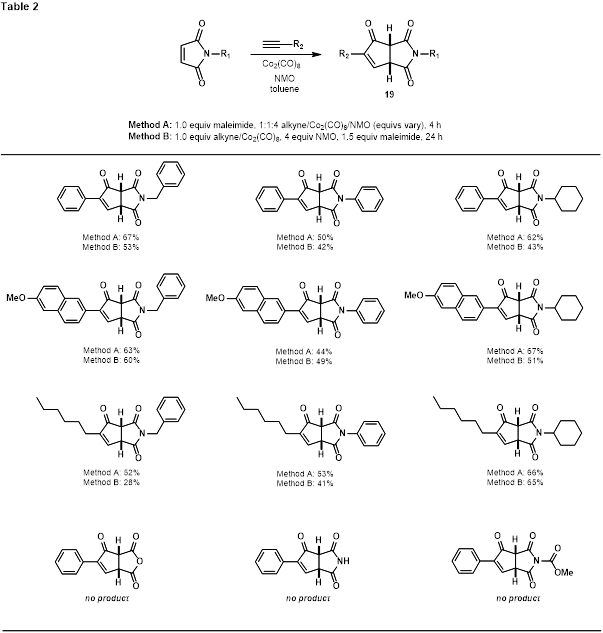
During the course of our studies, while attempting to engage the bridged silanes described in Scheme 1 in Pauson-Khand reactions, we discovered that N-benzylmaleimide reacted with Co2(CO)8 and terminal alkynes to produce fused cyclopentenone-pyrrolidine-2,5-diones (19, Table 2). Optimization of this transformation, notable due to the tendency of electron-deficient olefins to produce dienes rather than cyclopentenones when subjected to Pauson-Khand conditions,5 produced two protocols that were used in a brief survey of three alkynes and three N-substituted maleimines. Interestingly, N-H and N-acyl maleimides failed to deliver cyclopentenone product, as did maleic anhydride under these conditions.
This project has provided hands-on research opportunities in synthetic organic chemistry for a total of four undergraduate students and one Masters-level graduate student at UNC Wilmington. Two have gone on to PhD programs in chemistry, two have subsequently enrolled as Masters students in the Department of Chemistry and Biochemistry at UNC Wilmington, and one (an Honors student) is currently in the process of applying to PhD programs in chemistry. The PI is grateful to PRF for providing funding for this proposal, the first of his independent career. Through the many difficulties encountered while working on this project, both the students and the PI have benefited from the opportunity to make unexpected discoveries and pursue new investigations as circumstances dictated. The research program initiated with this PRF grant will continue and the results will be published in due course.
References:
1A. C. Stevens, B. L. Pagenkopf, Org. Lett. 2010, 12, 3658-3661.
2Rieke, R. D.; Xiong, H. J. Org. Chem. 1991, 56, 3109-3118.
3Franz, A. K.; Wilson, S. O. J. Med. Chem. 2013, 56, 388-405.
4Jiang, D.; Chen, Y. J. Org. Chem. 2008, 73, 9181-9183.
5Gibson, S. E.; Mainolfi, N. 2005, 44, 3022-3037.










