Reports: UNI655243-UNI6: Structure and Energetics of Calcium Carbonate Pre-Nucleation Clusters from Atomic-Scale Simulations: Development of a Novel Interatomic Potential
 printer friendly
printer friendlyIntroduction
The goal of this project is to develop a novel interatomic potential that will enable atomic-scale simulations for the structure and thermodynamics of calcium carbonate (CaCO3) pre-nucleation clusters in water. Our strategy is to extend the dissociable water potential of Garofalini and co-workers [1] to include parameters that describe the interactions between H2O, Ca2+, and CO32–. The reference data used for fitting the potential is generated from first principles molecular dynamics (FPMD) simulations, which makes no assumptions about the form of the interatomic interactions.
Our starting point is the CaCO3(aq) contact ion pair in water, which can be regarded as a basic building block for larger clusters of calcium carbonate. FPMD is employed to study the hydration structure around CaCO3(aq). We also compare with results derived from classical MD using one of the most recent CaCO3-water potentials [2]. This is a non-polarizable model potential that regards the carbonate as a rigid anion, and the water is modeled using the TIP4P-Ew [3]. As we discuss below, we find surprising, qualitative differences between results derived from the classical and FPMD simulations.
Classical molecular dynamics with the TIP4P-Ew-based potential
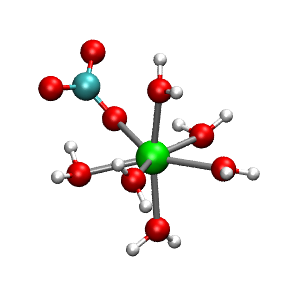
The simulation model for the classical MD is a 3D-periodic cubic cell with lattice parameter a = 12.43 Å, containing 62 water molecules and a single CaCO3(aq) contact ion pair. While classical MD can handle much larger system sizes, a small model is chosen here to enable direct comparison with the first principles MD (see below). Figure 1 (blue) shows the Ca-O radial distribution function, g(r), from the classical MD. The first peak at a distance of r = 2.35 Å corresponds to a well-defined inner coordination sphere of oxygens around the Ca2+, followed by a more weakly structured second shell at around r = 4.5 Å. Integrating the g(r) over the first peak yields seven oxygen atoms in the inner sphere. A more detailed examination of the complex reveals that one of the inner-sphere oxygens comes from the carbonate anion that is bound to the Ca2+ in a monodentate fashion, while the remaining six oxygens correspond to six waters of hydration (Figure 2).
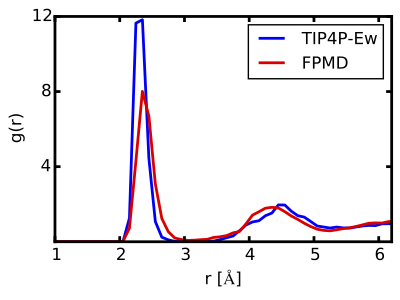
Figure 1: Ca-O radial distribution function, g(r). Results from classical MD using the TIP4P-Ew-based force field (blue) and from FPMD (red) are shown. While both models yield an inner coordination sphere at around r = 2.35 Å, the classical MD predicts a coordination number of seven while the FPMD gives six.
First principles molecular dynamics
The FPMD provides a qualitatively different picture of CaCO3(aq) hydration. In these simulations, the unit cell is the same as for classical MD described above. Interatomic forces are derived at each time step from Kohn-Sham density functional theory within the generalized gradient approximation of Perdew, Burke, and Ernzerhof (PBE) [4]. The result for the Ca-O radial distribution function is shown in Figure 1 (red). Similar to the classical MD, there is a well-defined first shell at r = 2.35 Å, followed by a more diffuse second shell at a closer distance of around r = 4.35 Å. The FPMD results differ markedly from the classical MD in the structure of the inner coordination sphere, which consists of six oxygens instead of the seven as predicted by classical MD. Moreover, the complex is dynamically switching between two different six-coordinate structures: i) a Ca2+ coordinated by a monodentate carbonate and five waters (Figure 3a), and ii) a Ca2+ coordinated by a bidentate carbonate and four waters (Figure 3b). This can be seen in the FPMD by monitoring an order parameter defined as the difference in distances between the two Ca2+-carbonate bonds (see Table of Contents graphic).
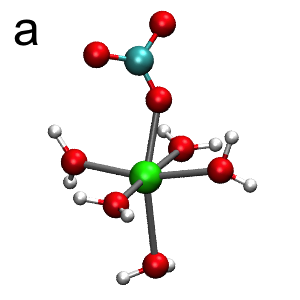
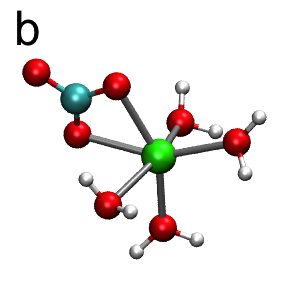
Figure 3: CaCO3(aq) hydration structures from FPMD. The color scheme is: Ca2+ (green), oxygen (red), carbon (cyan), and hydrogen (white). The Ca2+ complex interconverts between two distinct six-coordinate structures: (a) monodentate carbonate and five waters, and (b) bidentate carbonate with four waters.
Impact and next steps
The Petroleum Research Fund has been central to our efforts to study non-classical nucleation phenomenon. This grant has supported one undergraduate (BS) and one masters (MS) student, both of whom have recently graduated. We are in the process of recruiting and training new students for this project. The grant has also provided summer support for the PI plus travel to the Spring 2016 ACS National Meeting in San Diego, CA.
At this point, we have generated the reference FPMD data that will guide the development of a new CaCO3-water potential. The next step now is to extend the parameterization of the dissociable water potential [1] to include interactions with Ca2+ and CO32– in a way that is consistent with the structural characteristics and dynamical behavior seen in the FPMD.
References
[1] Mahadevan, T.S.; Garofalini, S.H. Dissociative Water Potential for Molecular Dynamics Simulations. J. Phys. Chem. B 2007, 111, 8919–8927. DOI: 10.1021/jp072530o
[2] Raiteri, P.; Gale, J.D.; Quigley, D.; Rodger, P.M. Derivation of an Accurate Force-Field for Simulating the Growth of Calcium Carbonate from Aqueous Solution: A New Model for the Calcite-Water Interface. J. Phys. Chem. C 2010, 114, 5997–6010. DOI: 10.1021/jp910977a
[3] Horn, H.W.; Swope, W.C.; Pitera, J.W.; Madura, J.D.; Dick, T.J.; Hura, G.L.; Head-Gordon, T. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J. Chem. Phys. 2004, 120, 9665. DOI: 10.1063/1.1683075
[4] Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. DOI: 10.1103/PhysRevLett.77.3865










