Reports: DNI455595-DNI4: Unraveling Heterocycle-Promoted Hydride Transfer Mechanisms for Energetically Efficient Fuel and Petrochemical Production
 printer friendly
printer friendlyOverview of Project:
Abstract:
Our society's future hinges on scientists and engineers being able to develop renewable fuels and chemicals. Recent studies have implicated aromatic N-heterocycles as playing key roles in energetically efficient conversion of CO2 into molecules such as CO, formate, and methanol, but the exact roles of these heterocycles are not well understood. Improved understanding would advance technologies for energetically efficient recycling of CO2 into renewable fuels and chemicals. We will use state of the art computational quantum chemistry to rigorously investigate electroreduction mechanisms starting from CO2, passing through formate and CO intermediates, up to methanol and/or other C-C bond formations that may be promoted by different classes of aromatic N-heterocycles. We will predict electrochemical conditions at which heterocycles facilitate energetically efficient hydride transfers as well as their reaction mechanisms. From this work, we aim to provide guidelines to advance the engineering of renewable fuel catalysts.
Progress:
Building from recent work done by our group, we have investigated the degree that redox reaction energetics and Pourbaix diagram isobars vary across a wide variety of ANH molecules. We used first-principles quantum chemistry to quantify the degree that nitrogen atoms, aromatic rings, and/or electron-withdrawing groups within molecular structures cause substantially less negative redox potentials (Figure 1). Although molecular redox potentials and pKas are sensitive to molecular structure, Pourbaix diagrams for nearly all of the reported ANH molecules have at least one triple point near the equilibrium potentials for CO2 reduction at the same pH (Figure 2). This signifies that ANH molecular moieties in general may be used to shuttle protons, electrons, and possibly hydrides at appropriate electrochemical conditions. Although experimental validation of ANH-promoted CO2 reduction mechanisms is still needed, calculated triple points are a theoretically sound descriptor for screening a molecule's (or material's) capability for catalyzing electrochemical reductions. Similar analyses can be performed straightforwardly with quantum chemical accuracy to assist in the identification of molecular co-catalysts for electrochemical reactions. This work was recently published in J. Phys. Chem. A.
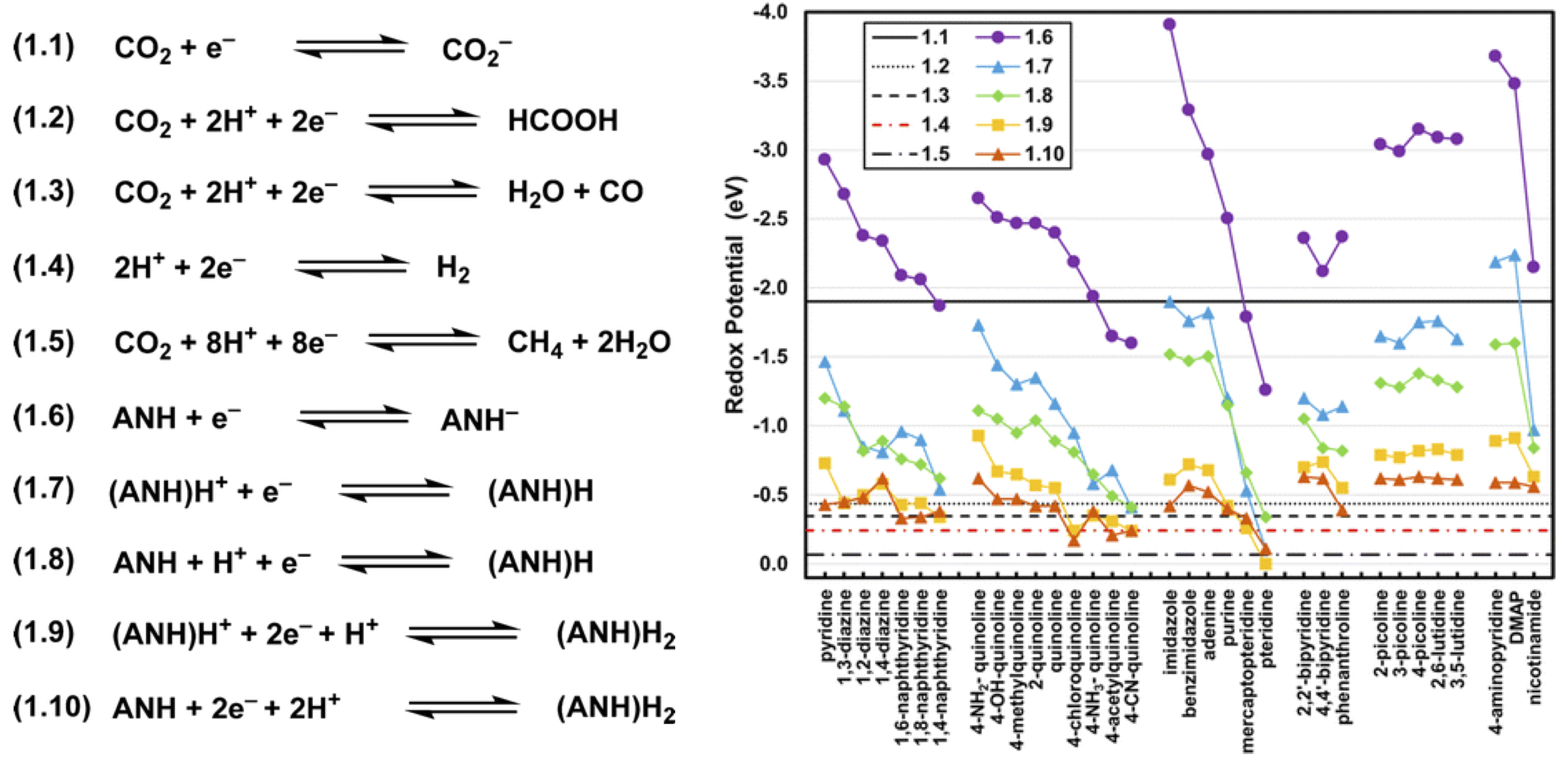
Figure 1. Redox potentials for 20 ANH molecules calculated at 0 pH. Lines correspond to redox processes. DMAP corresponds to N,N-dimethylaminopyridine.
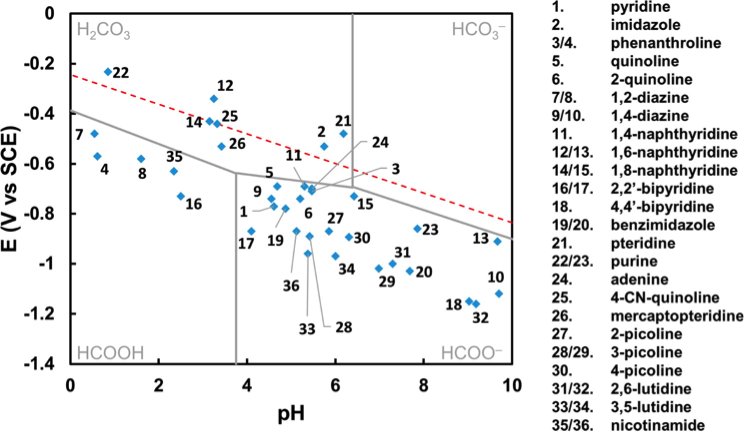
Figure 2. Compiled Pourbaix diagram triple point conditions for all considered molecules. The red dashed line corresponds to hydrogen evolution reaction. Gray lines correspond to the CO2 equilibrium products and standard redox potentials in aqueous environments.
We have also carried out an extensive atomistic thermodynamics study of aromatic N-heterocycle ligands and Ru complexes. We have determined pKas, SRPs, and hydricities for these molecules and inorganic complexes considering up to two proton and two electron transfers to determine which states would be thermodynamically stable under electrochemical CO2 reduction operating conditions. As found previously with our other studies, the molecules and complexes were found to have several metastable states that include up to multiple proton and electron transfers. These intermediate states in principle may open unexpected pathways for CO2 hydrogenation such as biomimetic proton and hydride shuttling. While the pKas and 1e− SRP for [Ru(bpy)3]2+ and [Ru(phen)3]2+ suggest high energy barriers for protonation and reduction of those complexes, it remains unclear how [Ru(phen)3]2+ would participate in CO2 reduction in light of the instability of PyH+ radicals. However, the [Ru(phen)2(ppy)]2+ system used by MacDonnell and coworkers for CO2 reduction is a much more likely candidate for participating in CO2 reduction mechanisms involving proton and electron shuttling. In this system, computational predictions indicate that proton and hydride shuttling pathways are thermodynamically feasible and should be considered for energetically efficient CO2 reduction processes involving [Ru(phen)2(ppy)]2+. This work is currently in press at Dalton Trans.
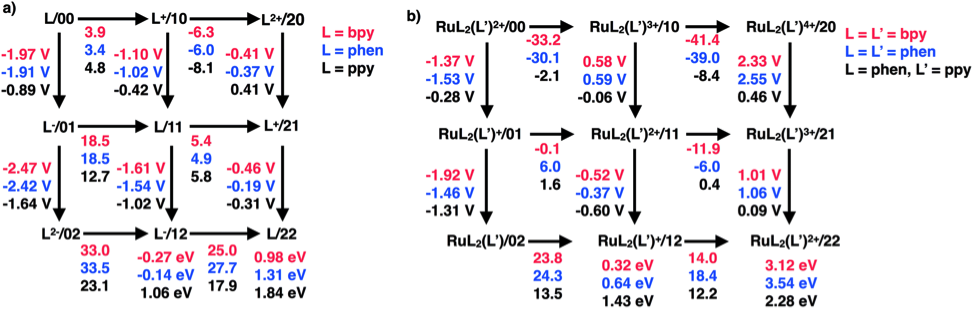
Figure 3. 'Square plots' denoting calculated SRPs, pKas, and hydricities for (a) ligands and (b) Ru complexes. The first and second digits in the labels following the '/' correspond to the number of protons and electrons added to the initial species from the species listed in the top left of each figure.
In other efforts we are modeling nanocarbon materials for their propensity to participate in energetically efficient hydride transfer mechanism. A submitted manuscript for this work is currently under review in the journal, Carbon.
A central component of our grant was to model electrochemical molecular properties of aromatic N-heterocycles interacting with electrode surfaces. We originally proposed to model these systems using surface cluster models embedded in dielectric continuum models. We have carried out a benchmarking study reporting the accuracy of this approach compared to existing computational models. We conclude that continuum solvation models obtained from periodic slab calculations and surface cluster models are generally similar and robust, and they provide meaningful insight of solid/liquid interfaces as long care is taken to ensure that surface clusters do not exhibit structural artifacts and error cancellation schemes are used. Future work studying the energetic contributions of explicit molecular species at a solid/liquid interface will be pursued in future work. A submitted manuscript for this work is currently under review in the journal, Mol. Simul.
Future Plans:
We are now investigating modeling procedures to accurately describe the role of local solvation environments in regulating the atomic scale nature of hydrogenation mechanisms. The modeling approach we originally proposed was later determined to be inappropriate because it did not account for critical contributions arising from configurational entropies from solvents. To address this, we have worked on modeling reaction pathways in aqueous solution using different computational methods. We will establish what level of computational sophistication is needed to accurately model hydride transfers in realistic electrolytes. The results for this effort currently ongoing, but our findings will be reported in the next project report period.










