Reports: DNI355302-DNI3: Mechanism and Scope of Bis(imino)pyridine Manganese-Catalyzed Hydrosilylation
 printer friendly
printer friendlyWe have had success investigating three different aspects of manganese-mediated hydrosilylation. Our efforts to elucidate the mechanism of bis(imino)pyridine (or pyridine diimine, PDI) manganese-catalyzed hydrosilylation, determine the scope of this transformation, and modify the ligand to enhance this scope are described below.
Mechanism of Hydrosilylation. We previously reported that PDI manganese precatalyst, (Ph2PPrPDI)Mn (1) (Fig. 1), is active for the hydrosilylation of ketones and dihydrosilylation of esters. Under stoichiometric and catalytic conditions, we found that adding PhSiH3 to (Ph2PPrPDI)Mn results in partial conversion to a diamagnetic hydride complex, (Ph2PPrPDI)MnH (2) (Fig. 1). Independent preparation of 2 from (Ph2PPrPDI)MnCl2, followed by single-crystal X-ray diffraction analysis, has revealed this complex to possess a capped trigonal bipyramidal geometry. When 2,2,2-trifluoroacetophenone is added to 1, radical transfer has yielded (Ph2PPrPDI·)Mn(OC·(Ph)(CF3)) (3) (Fig. 1), which undergoes intramolecular C-C formation to form the respective Mn(II) dimer, [(μ-O,Npy-4-OC(CF3)(Ph)-4-H-Ph2PPrPDI)Mn]2 (4) (Fig. 1). Upon finding 3 to be inefficient and 4 to be inactive, we have conducted kinetic trials to shed light on the mechanisms of 1- and 2-mediated hydrosilylation. Varying the concentrations of 1, substrate, and PhSiH3 has revealed a first order dependence on each reagent. A kinetic isotope effect (KIE) of 2.2 has been observed for the 1-catalyzed hydrosilylation of 2,4-dimethyl-3-pentanone, suggesting Si-H(D) bond activation contributes to the rate determining step. In contrast, a KIE of 4.2 has been observed when using 2 as the catalyst, suggesting an alternate mechanism of hydrosilylation. Although kinetic trials revealed 1 to be the more active precatalyst for carbonyl hydrosilylation, a concurrent 2-mediated pathway is efficient for ester hydrosilylation. Considering our observations, 1-catalyzed hydrosilylation is believed to proceed through a modified Ojima mechanism, while 2-mediated hydrosilylation occurs via insertion.
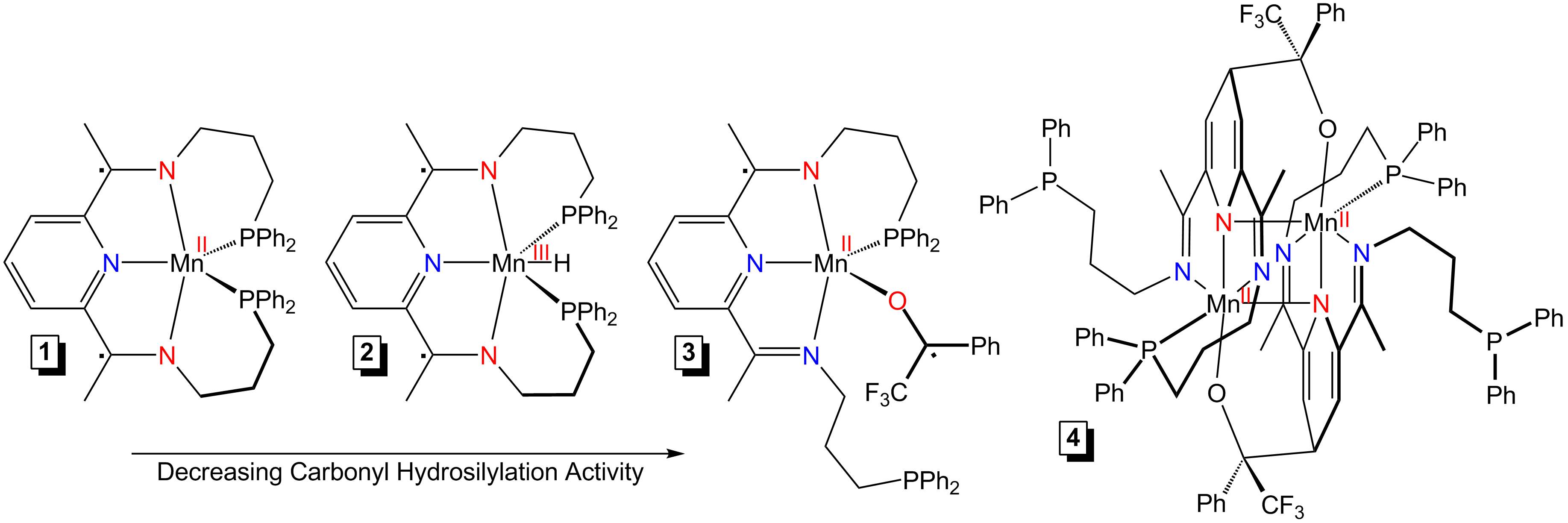
Fig. 1. Manganese compounds relevant to carbonyl hydrosilylation.
Hydrosilylation Scope. We have also been working to demonstrate an expanded scope for 1-mediated hydrosilylation. Adding a neat mixture of benzaldehyde and PhSiH3 to 0.1 mol% 1 at 25 °C has resulted in complete conversion to silyl ethers within 2 min and Treatment with 10% aq. NaOH allows for isolation of benzyl alcohol following extraction. Thirteen different aldehydes have been hydrosilylated under these conditions and 1 has been found to tolerate a range of functionalities (Fig. 2). Halide-substituted benzaldehydes were converted to silyl ethers without issue except for 4-iodobenzaldehyde, which has showed 32% conversion. The cyano-group of 4-cyanobenzaldehyde and nitro-group of 4-nitrobenzaldehyde remain unreduced during the reaction. Electron-donating methyl and methoxy substituents have not interfered with conversion efficiency and hydrosilylation rates are not affected by the heterocyclic functionalities of pyridine-3-carboxaldehyde and furfural. For unsaturated aldehydes such as 3-cyclohexene-1-carboxaldehyde and citral, the aldehyde functionality is reduced and the olefin is not, consistent with previous attempts at alkene hydrosilylation.
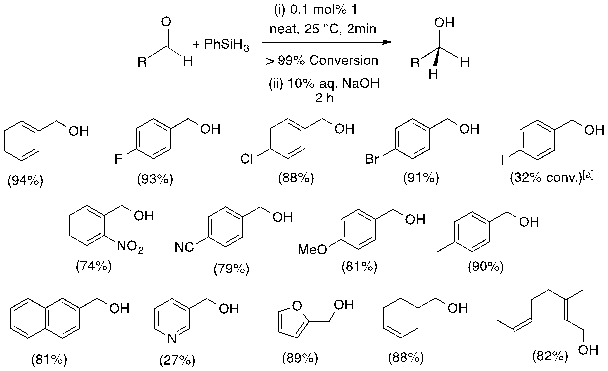
Fig. 2. Aldehyde hydrosilylation scope using 1.
The hydrosilylation of benzaldehyde, 2-naphthaldehyde, and 4-fluorobenzaldehyde has been conducted separately using 0.01 mol% catalyst loading under identical conditions and each substrate is transformed into silyl ethers with TOFs of up to 4,950 min-1. The hydrosilylation of 4-nitrobenzaldehyde has proceeded with a maximum TOF of 3,600 min-1 (Fig. 3). Hydrolysis of the silyl ethers followed by workup yields pure benzyl alcohol (92%), 2-naphthylmethanol (83%), and 4-fluorobenzyl alcohol (87%). After exploring activity and functional group tolerance, 1 was tested for longevity under higher substrate loading. To determine maximum TON for 1-catalyzed aldehyde hydrosilylation, a mixture of 10,000 equivalents PhSiH3 and PhCHO was added to 1 and repeated 4 times (total of 50,000 equivalents) at 15 min intervals. Analysis of the resulting mixture revealed 67% conversion, indicating a maximum TON of 31,500.

Fig. 3. Optimization of 1-catalyzed aldehyde hydrosilylation.
Knowing that 1 can cleave ester C-O bonds, the hydrosilylation of formates using 1 has also been investigated. When a neat equimolar mixture of methylformate (or ethylformate) and PhSiH3 is added to 0.02 mol% 1, an exothermic reaction occurs with >99% substrate conversion in 15 min. Attempts to hydrolyze the resulting silyl ethers to isolate the corresponding alcohols by fractional distillation has not allowed for adequate separation, although choosing higher molecular weight formates has. Adding an equimolar quantity of benzyl formate and PhSiH3 to 0.02 mol% 1 results in vigorous bubbling. Catalyst deactivation after 15 min has revealed complete consumption of starting formate. Hydrolysis using 10% aq. NaOH, followed by extraction and evaporation, has afforded pure benzyl alcohol in excellent yield (88%). Similarly, phenylethyl formate, isoamyl formate, and hexyl formate have been reduced to the corresponding alcohol. Interestingly, geranyl formate and p-anisyl formate have displayed <2% conversion after 1 h of stirring at 25 °C. Unlike acetates, the TOFs of formate reduction are unaffected upon varying the ester group. A TOF of 330 min-1 (based on substrate conversion) has been achieved for each substrate, which is the highest TOF for first row metal catalyzed formate hydrosilylation known to date. We are continuing to investigate the hydrosilylation of carboxylic acids, imines, and nitriles.
Chelate Modification. Heating (THF)2MnCl2 in the presence of the pyridine-substituted ligand, PyEtPDI, has afforded the respective dihalide compound, (PyEtPDI)MnCl2. Reduction of this compound using Na/Hg results in deprotonation of the chelate to yield the bis(enamide)tris(pyridine)-supported product, (κ5-N,N,N,N,N-PyEtPDEA)Mn (5), rather than a compound that is formally zerovalent. This compound has been structurally characterized and found to possess an intermediate spin Mn(II) center by Evans Method and EPR spectroscopy. Even though it lacks chelate redox-activity, 5 has been found to hydrosilylate aldehydes and ketones with excellent TOFs of 2,475 min-1 under neat conditions. This optimization has allowed for isolation of the respective alcohols, and in two cases, the partially reacted silyl ethers, PhSiH(OR)2 (R = Cy and CH(Me)(nBu)). The aldehyde hydrosilylation activity observed for 5 renders it one of the most efficient first row transition metal catalysts for this transformation reported to date. Unfortunately, this compound has not been found to mediate alkene, alkyne, or carbon dioxide hydrosilylation, which remain the primary limitations of 1 and 2. We are actively investigating ligand modifications that can further improve the scope of Mn-mediated hydrosilylation.
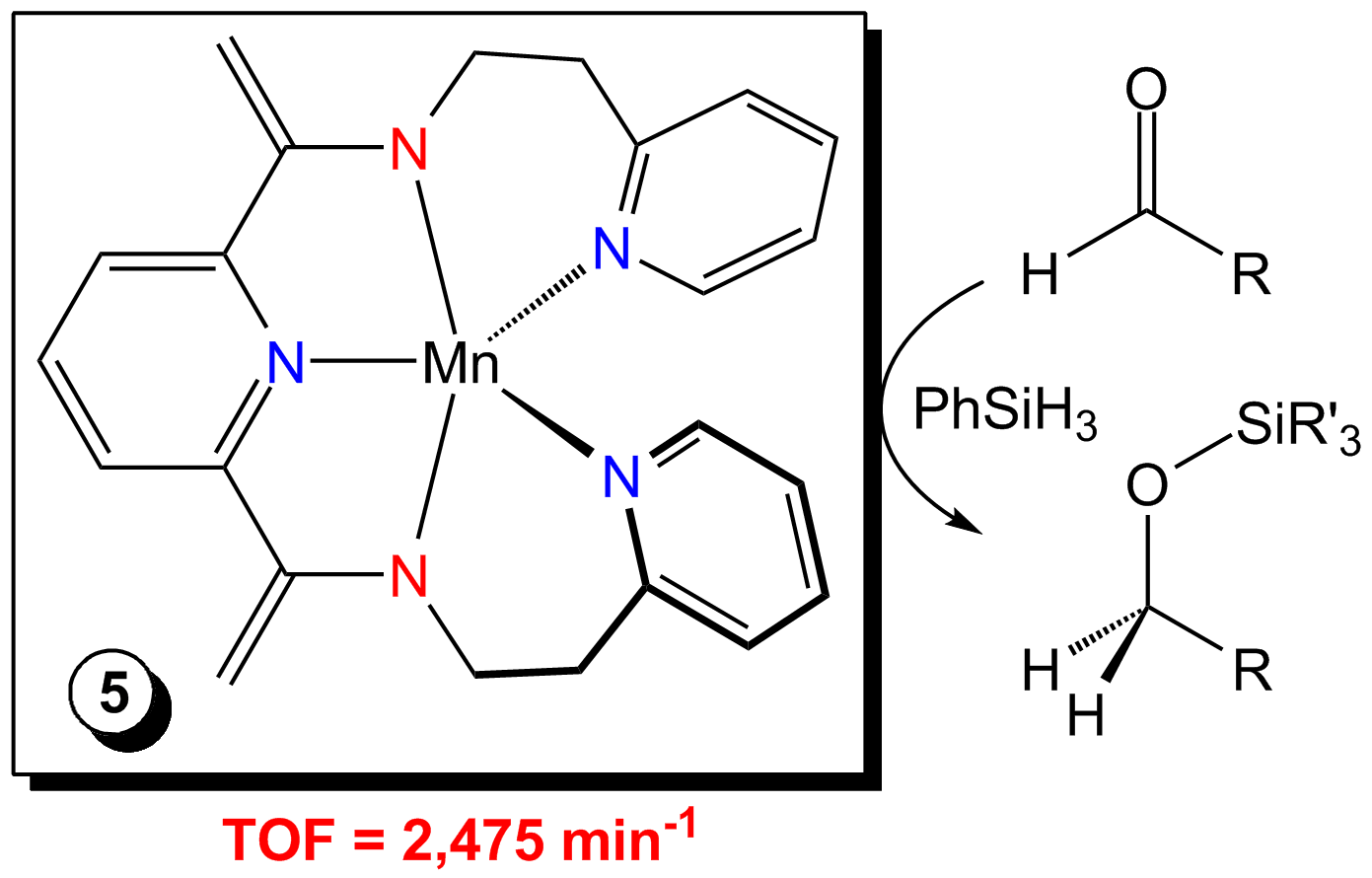
Fig. 4. Chelate deprotonation affords an efficient Mn hydrosilylation catalyst.










